Research ArticleCardiologyGenetics
Open Access | ![]() 10.1172/jci.insight.196933
10.1172/jci.insight.196933
Extracellular matrix alterations in chronic ischemic cardiomyopathy revealed by quantitative proteomics
Kevin M. Buck,1 Holden T. Rogers,1,2 Zachery R. Gregorich,2,3 Morgan W. Mann,4 Timothy J. Aballo,2,5 Zhan Gao,2 Emily A. Chapman,1 Andrew J. Perciaccante,1,2 Scott J. Price,2 Ienglam Lei,6 Paul C. Tang,6,7 and Ying Ge1,2,8
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Buck, K. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Rogers, H. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by
Gregorich, Z.
in:
PubMed
|
Google Scholar
|

1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Mann, M. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Aballo, T. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Gao, Z. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Chapman, E. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Perciaccante, A. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Price, S. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Lei, I. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Tang, P. in: PubMed | Google Scholar
1Department of Chemistry,
2Department of Cell and Regenerative Biology,
3Department of Animal and Dairy Sciences,
4Department of Medicine, and
5Molecular and Cellular Pharmacology Training Program, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Department of Physiology and Biomedical Engineering and
7Department of Cardiac Surgery, Mayo Clinic, Rochester, Minnesota, USA.
8Human Proteomics Program, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Ying Ge, 1111 Highland Ave., WIMR II 8551, Madison, Wisconsin 53705, USA. Email: ying.ge@wisc.edu.
Find articles by Ge, Y. in: PubMed | Google Scholar
Published September 30, 2025 - More info
JCI Insight. 2025;10(21):e196933. https://doi.org/10.1172/jci.insight.196933.
© 2025 Buck et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
-
Results
Global proteomics analysis of failing ICM and nonfailing donor tissues. To identify differences in the composition of the ECM in failing ICM versus nonfailing donor myocardium, we optimized our cardiac tissue extraction protocol to maximize solubilization of ECM proteins. Our group had previously demonstrated the efficacy of using the photocleavable surfactant, Azo, alone and in combination with a decellularization step using a highly concentrated salt, such as NaCl, in a sequential extraction (15, 22). However, this approach had not been applied to fibrotic, dense muscle tissues, such as human cardiac tissue. Prior studies have reported the improved performance of LiCl over NaCl in the extraction of proteins from muscle tissues (23), and our group has successfully used LiCl to extract myofibrillar proteins from human heart tissue (24). Thus, we employed sequential extractions first with LiCl followed by Azo-containing buffer to effectively decellularize tissue and solubilize ECM proteins.
To gain insight into differences in the composition of the ECM in end-stage ICM, this optimized sequential protein extraction protocol was employed to decellularize tissue and solubilize ECM proteins from the apex of the LV myocardium of failing ICM (n = 16) and nonfailing donor (n = 16) hearts (Figure 1A). The LiCl and Azo extracts (64 total samples; n = 32 ICM, n = 32 control) were subjected to analysis by MS-based proteomics. The total ion chromatograms from each extract across cohorts and normalized intensities following searching demonstrated consistency between biological replicates (Supplemental Figures 1 and 2; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.196933DS1).
 in failing ICM and nonfailing donor heart tissues.") Figure 1
Figure 1Azo-enabled ECM proteomics method yields reproducible coverage of the matrisome (the collection of proteins in the ECM) in failing ICM and nonfailing donor heart tissues. (A) Schematic representation of the workflow for MS-based proteomic analysis of protein extracts from human ICM and donor patient myocardial tissue. Soluble proteins are depleted in the first LiCl extraction (LiCl) followed by solubilization of “insoluble” ECM proteins by extraction with buffer containing the photocleavable surfactant, Azo. (B) CVs of protein intensities across extracts from failing ICM and nonfailing donor tissues. The median CV of protein intensities was below 5% for extracts from both groups, indicating high quantitative reproducibility. CVs were calculated by (SD/mean intensity) × 100% for each quantified value in each replicate. (C) Upset plot and bar graph (inset) showing more than 6,000 unique protein identifications overall, as well as the degree of overlap between groups. (D and E) Simplified depiction of major categories of ECM proteins (per MatrisomeDB, ref. 25) (D) and breakdown of the number of proteins in each category identified in this study (E). The inner circle shows the total number of proteins in each category while the outer circle indicates those that were identified herein. Black segments correspond to proteins present in MatrisomeDB (25) that were not identified in human ICM and donor myocardial tissue.
For each of the extracts and disease conditions, quantitative reproducibility was assessed by plotting the coefficients of variation (CVs) for the protein-level normalized label-free quantified (LFQ) intensities (Figure 1B). All groups showed median CV values of less than 5%. In addition, normalized LFQ intensities were plotted for each replicate against the others within each of the 4 sample groups and showed an average Pearson’s correlation coefficient of r = 0.948. For ICM samples, the average r values for the LiCl and Azo extracts were 0.936 and 0.93, respectively, while for nonfailing controls they were 0.966 and 0.96 (Supplemental Figure 3).
Following database searching of raw mass and tandem mass spectra, data were filtered to retain only genes with 8 or more valid quantified values in at least 1 of the conditions, resulting in the identification of a total of 6,125 unique protein groups across both extracts. After a second filtration step wherein proteins with more than 50% imputed values were removed, 6,118 unique protein groups remained. Overall, more protein groups were quantified in the LiCl extract than the Azo extract, though Azo exhibited a greater degree of overlap between ICM and control donor tissues, suggesting better consistency in detecting shared proteins (Figure 1C). Notably, when comparing the protein intensities across all extracts, we found that major fibrillar proteins, such as type 1 collagen, fibronectin, and elastin, were significantly more abundant in Azo extracts, showing our method effectively enriched insoluble ECM proteins (Supplemental Figure 4). To assess the effectiveness of ECM extraction, the total number of ECM proteins in all extracts was identified according to information in MatrisomeDB (25) and plotted according to their category. These categories consist of collagens, proteoglycans, glycoproteins, ECM regulators, secreted proteins, and ECM-affiliated proteins (Figure 1D). These results demonstrated that our method was effective at extracting all categories of ECM proteins, with particularly high coverage of glycoproteins, of which our dataset captured approximately 55% of those in the database (Figure 1E).
Alterations in failing human ICM myocardium revealed by global proteomics. Principal component analysis (PCA) of the Azo extract showed clear separation between failing ICM and nonfailing donor samples along PC1, which accounted for 33.34% of the total variance (Figure 2A). Similar clustering by condition was observed in the PCA results of the LiCl extract, though the separation explained a smaller proportion of the variance (Supplemental Figure 5). In general, ICM samples had less cohesion in PCA clustering, which alongside lower Pearson’s correlation coefficients can be explained by the relatively higher degree of heterogeneity in the ICM tissues compared with controls. This can likely be explained by differences in factors such as prior MI status, treatments, and comorbid conditions like diabetes (Supplemental Table 1). Differential expression analysis was performed by a limma test, yielding 313 differentially expressed protein groups in the Azo extract (of 2,489 quantified protein groups) and 826 differentially expressed protein groups in LiCl. These were sorted in descending order of adjusted P value, and then the top 300 results were plotted in a hierarchical heatmap and clustered by k-means, revealing a distinct pattern in protein abundance in failing ICM compared with nonfailing donor myocardium (Figure 2B).
 Figure 2
Figure 2Global proteomics results from ECM-enriched extract reveals alterations in failing ICM versus nonfailing donor myocardium. (A) PCA shows clustering and clear separation of failing ICM and nonfailing donor group replicates. The ellipse represents the 95% confidence interval of the data assuming a multivariate normal distribution. (B) Heatmap of Z-scored protein intensities (top 300 most significant differentially expressed) shows clusters (k-means) of proteins up- and downregulated in failing ICM versus nonfailing control myocardium. (C) Bubble plots showing GO terms enriched among protein clusters up- (black) and downregulated (gray) in end-stage failing ICM compared with nonfailing control. Enriched cellular component terms (left) highlight up- and downregulation of ECM and mitochondrial proteins, respectively, in failing ICM compared with nonfailing control myocardium. Similarly, biological process terms (right) enriched among proteins upregulated in the failing ICM myocardium pertained to ECM organization and complement activation while those enriched among downregulated proteins were concerned with metabolic processes. FDR is P value–corrected by the Benjamini-Hochberg method, done automatically by STRING.
Differentially expressed proteins broadly clustered into 2 groups: those that were up- and those that were downregulated in failing ICM samples relative to nonfailing controls. Gene Ontology (GO) analysis was performed for differentially expressed proteins, and the GO terms, associated proteins, enrichment strength, and false discovery rate (FDR) are reported in Supplemental Table 4. GO analysis of the 225 proteins upregulated in failing ICM versus nonfailing donor myocardium showed unambiguous association with the ECM (GO:0031012; FDR = 2.83 × 10–43) and its functions, including ECM organization (GO:0030198; FDR = 2.51 × 10–18), wound healing (GO:0042060; FDR = 4.59 × 10–12), and regulation of complement activation (GO:0030449; FDR = 1.68 × 10–10). Other enriched terms were related to blood coagulation (GO:0007596; 9.83 × 10–11), platelet degranulation (GO:0002576; FDR = 3.9 × 10–11), proteolysis (negative regulation of endopeptidase activity; GO:0010951; 6.92 × 10–10), and cell adhesion (GO:0007155; FDR = 1.47 × 10–5). The 86 downregulated proteins were associated with the mitochondrion (GO:0005739; FDR = 5.48 × 10–11) and oxidative phosphorylation (GO:0006119; FDR = 1.51 × 10–9), underscoring the established link between mitochondrial dysfunction and HF (3, 26) (Figure 2C). GO analysis from the LiCl extract exhibited nearly identical trends, with more terms related to cytosolic proteins attributable to the role of this step in decellularization of tissues (Supplemental Figure 6).
Cardiac ECM remodeling in end-stage HF from ICM. Global proteomics results showed that many of the detected ECM proteins (68 out of the total 315 ECM proteins detected) were upregulated in failing ICM compared with nonfailing donor heart tissue. We contextualized these findings using information in MatrisomeDB (25) to outline categories by which to observe matrisome changes in ICM. In total, 315 ECM proteins were identified from the raw data search, which reduced to 286 after all filtering steps, consisting of 89 glycoproteins (46.6% of database), 18 collagens (40%), 14 proteoglycans (38.9%), 37 ECM-affiliated proteins (22.9%), 87 ECM regulators (35%), and 41 secreted proteins (12.4%). To visualize the categorical ECM differences in ICM compared with control hearts, we plotted the quantile function (0.95% confidence interval) of the log2 fold changes of differentially expressed ECM proteins across each matrisome category (Figure 3A). Categories that were considered significantly upregulated in failing ICM versus nonfailing control tissues had a mean fold change (± the confidence interval) greater than 0, indicating that glycoproteins (P value = 5.23 × 10–11), ECM regulators (P value = 2.29 × 10–5), proteoglycans (P value = 1.62 × 10–5), and collagens (P value = 4.02 × 10–5) showed significantly higher abundance in Azo of the ICM samples categorically. The same categories were significant in LiCl with the exceptions of ECM-affiliated proteins and collagens, the latter of which we can assume is attributable to increased collagen solubilization by the Azo surfactant. Table 1 summarizes the P values and log2 fold changes for several categories of ECM proteins significantly upregulated in the failing ICM versus nonfailing control myocardium based on the Azo data. These categories, though broadly altered, are functionally heterogeneous, so subsequent sections will highlight individual proteins to provide more detailed interpretation of these changes. The individual proteins from the table are plotted in categories as box plots showing individual replicates in Supplemental Figures 7–10.
 Figure 3
Figure 3ECM remodeling in end-stage HF from ICM. (A) Dot plot showing categorical log2 fold change in the intensities of matrisome proteins. Mean and SD of log2 fold changes for all ECM proteins in each extract and category were plotted. Overall, core matrisome proteins (collagens, proteoglycans, and glycoproteins), as well as ECM regulators, were significantly upregulated in failing ICM LV tissue. Means were tested for significance using Student’s 2-tailed t test, and the 97.5% confidence interval was computed. (B) Volcano plot showing select proteins up- and downregulated in failing ICM versus nonfailing donor myocardium. To be considered significant, a log fold change > 0.6 (>1.5-fold) and an adjusted P < 0.05 (–log p-adj > 1.3) were both required. (C) Core collagens, ECM regulators, glycoproteins, and proteoglycans show increased expression in ICM. Box plots show the interquartile range, median (line), and minimum and maximum (whiskers). Significance testing was performed using limma. Then P values were adjusted by independent hypothesis weighting and plotted as follows: *** ≤ 0.001. (D) Clustered heatmap and corresponding bubble plots showing the results of GO analysis using the list of ECM proteins upregulated in failing ICM myocardial tissue. In the first cluster, biological processes (top) relate to carbohydrate metabolism with molecular functions (bottom) of binding and matrix structural properties. The second cluster pertains more to matrix regulation by growth factors and peptidases. FDR is P value–corrected by the Benjamini-Hochberg method, done automatically by STRING.
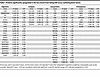 Table 1
Table 1Proteins significantly upregulated in the Azo extract from failing ICM versus nonfailing donor hearts
Plotting the log2 fold changes and adjusted P values (with thresholds of 0.6 and 0.05, respectively) revealed that the collagen and elastin cross-linking enzyme LOXL1 was the most highly upregulated protein in the failing ICM, with the lowest P value (3.57 × 10–31) and second-largest log2 fold change (2.60) (Figure 3, B and C). In other disease models including idiopathic pulmonary fibrosis and liver cirrhosis, LOXL1 was shown to be stimulated by pro-fibrotic TGFB to increase cross-linking, and its knockdown protects from fibrosis and reduces expression of pro-fibrotic metalloproteases and collagens (27). The next most significantly upregulated protein based on P value was the adhesive ECM glycoprotein FBLN1 (P value = 4.6 × 10–15), which also had the fourth-highest log2 fold change overall in the Azo extract (Figure 3, B and C). During cardiac development, FBLN1 promotes ADAMTS-1 (a disintegrin and metalloproteinase with thrombospondin motifs 1) cleavage of chondroitin sulfate proteoglycan VCAN (28). VCAN accumulation has been previously reported in failing hearts and shown to be reduced upon treatment with beta blockers regulated by ADAMTS family proteins (18). Consistent with this, we observed significantly higher VCAN expression in the failing ICM compared with nonfailing donor tissue (P value = 3.22 × 10–4) (Figure 3, B and C). These findings suggest that the marked increase in FBLN1 observed in failing ICM may serve as a protective mechanism, potentially compensating for elevated VCAN expression by stimulating ADAMTS-mediated proteolysis.
To further probe the pathways underlying these ECM differences in ICM, we performed hierarchical clustering of the ECM proteins significantly upregulated in failing ICM compared with nonfailing donor myocardium, again with 2 k-means clustering (Figure 3D). In cluster 1 we observed many terms related to glycosaminoglycans (GAGs), including GAG metabolism (GO:0030203; FDR = 6.13 × 10–8), catabolism (GO:0006027; FDR = 1.05 × 10–7), and sulfur compound metabolic process (GO:0044273; FDR = 1.44 × 10–6) relating to GAG sulfation. Of note, there were several terms related to cartilage development (GO:0051216; FDR = 1.05 × 10–7). Evidence shows similar hierarchical organization and regulation of ECM in both heart and cartilaginous connective tissues, including regulatory pathways involving aggrecan (ACAN) and tenascins (TNC and TNXB), which we found were significantly upregulated in ICM tissues (29, 30). The GO functional enrichment analysis returned terms primarily related to binding — collagen binding (GO:0005518; FDR = 1.26 × 10–9), GAG binding (GO:0005539; FDR = 1.33 × 10–12), integrin binding (GO:0005178; FDR = 1.40 × 10–4), ECM binding (GO:0050840; FDR = 1.30 × 10–3), and calcium ion binding (GO:0005509; FDR = 2.32 × 10–6) — but also had terms related to specific matrix structural elements. ECM structural constituents conferring tensile strength (i.e., resistance to stretching) were all collagens (COL1A2, COL3A1, COL6A2, COL6A3, COL12A1, COL14A1; GO:0030020; FDR = 2.93 × 10–8), while those conferring compression resistance were proteoglycans (DCN, VCAN, LUM, BGN, PRELP; GO:0030021; FDR = 1.28 × 10–7). Cluster 2 terms overlapped greatly with the generally upregulated protein groups, with major categories such as coagulation (e.g., negative regulation of blood coagulation; GO:0030195; FDR = 4.66 × 10–6) and proteolysis (GO:0052547; FDR = 2.86 × 10–7). Functional terms make it clear these trends relate to the SERPIN family of serine protease inhibitors (GO:0004867; FDR = 8.18 × 10–6) and matrix metalloproteinases, MMP9 in particular. Growth factor signaling also appeared as an enriched term, which is consistent with the role of TGFB in promoting ECM remodeling and fibrosis in ICM (31, 32).
Pathway analysis revealed TGFB signaling drives ECM remodeling in end-stage ICM. We next performed functional annotation clustering using Database for Annotation, Visualization and Integrated Discovery (DAVID) (33) to group similar annotations and clarify broader trends in the differentially expressed proteins in ICM tissues (Supplemental Table 4). Clusters were ranked by enrichment score, a geometric mean of P values for terms within a cluster, and using the differentially expressed proteins in the Azo extract, we observed 20 clusters wherein the top term’s Benjamini-Hochberg–adjusted P value was significant (<0.05). The top 2 clusters (enrichment scores = 22.77 and 16.19) unsurprisingly contained Kyoto Encyclopedia of Genes and Genomes (KEGG) and GO terms relating to ECM components, though the second more generically related to ECM components and the first had annotations of secreted proteins and glycoproteins. The next highest clusters were related to blood coagulation (enrichment score = 6.13), cell adhesion (5.12), calcium-binding epidermal growth factor domains (3.49), and SERPINs (3.48). Clusters 9 and 10 reflected complement pathways/innate immunity (2.97) and immunoglobulin binding (2.92), and cluster 12 reflected small leucine-rich proteoglycans (SLRPs), such as DCN, LUM, BGN, and PRELP (2.58).
To provide the context of protein-protein interactions into the pathway analyses, we used the R package pathfindR (34) to perform an active subnetwork search in a protein interaction network, then performed pathway enrichment using these networks. A P value threshold of 0.05 was used for significance, and active subnetwork search was iterated 10 times, with the lowest P value among iterations used to determine the overall significance of the term. Using KEGG annotations, 51 terms were significantly enriched from Azo using this approach, and the terms with the largest fold changes were complement and coagulation cascades (lowest P value = 1.06 × 10–21), oxidative phosphorylation (P value = 3.04 × 10–26), cardiac muscle contraction (P value = 3.87 × 10–10), and ECM-receptor interaction (P value = 3.28 × 10–11) (Figure 4A).
 Figure 4
Figure 4Pathway analysis implicates the TGFB signaling network in altered ECM composition in the myocardium of individuals with end-stage failing ICM. (A) Heatmap of agglomerated Z-scores for KEGG pathway terms across samples showing upregulation of pathways related to focal adhesions and ECM-receptor interactions. In addition, these changes are accompanied by increased regulation of the cytoskeleton and decreased expression of metabolic and contractile proteins. Term changes are visualized for the ECM-enriched extract. (B) Term-protein network generated using input data from both extracts demonstrates connections between pathways via specific proteins. In general, adhesive glycoproteins (thrombospondins, tenascins, fibronectin, vitronectin) and proteoglycans (decorin) connect receptor processes to focal adhesion and regulation by protein digestion and TGFB pathways.
From GO terms (using all terms from cellular component, biological process, and molecular function), 92 entries were deemed significant. Significant GO terms were largely in agreement with the previous GO results, though the fold enrichment provided for each term provided new context for particularly large changes, including the 61-fold enrichment for ECM structural constituents conferring compression resistance (P value = 2.38 × 10–2), a category containing mainly small leucine-rich proteoglycans. Notably, integrin activation (P value = 2.72 × 10–5) and integrin-mediated signaling pathways (P value = 3.39 × 10–5) were also significant, displaying a 24-fold and a 10-fold enrichment, respectively. Hierarchical clustering of the enriched terms linked ECM organization to integrin binding (Supplemental Figure 11), and plotting individual proteins showed FN1, APOE, and EFEMP2 linked those terms as well as endoplasmic reticulum lumen, heparin binding, and peptide cross-linking (Supplemental Figure 12).
Reactome searches yielded 134 significant terms, of which the majority with the lowest P values were related to inflammation, hemostasis, and dysregulated electron transport. Insulin-like growth factor binding was previously shown as a significant term from GO analyses, and a similar Reactome annotation, Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs), provided a more specific description of the pathway with an expanded list of relevant proteins. Hierarchical clustering showed multiple distinct groups relating to ECM organization, collagen biosynthesis, and ECM degradation (Supplemental Figure 13). In addition, clusters involving syndecan/nonintegrin cell-surface signaling, integrin MAPK signaling, complement cascades, and Toll-like receptor signaling were all enriched. Fibrinogens linked Toll-like receptors and clot formation to ECM signaling, while FN1 again spanned categories of ECM organization and cell surface signaling by integrins and syndecans (Supplemental Figure 14). LiCl extract analysis displayed the same ECM-related terms as in Azo but also captured TGFB signaling (P value = 2.2 × 10–2). Plotting relevant ECM-related terms, there was clearly a substantial degree of overlap between TGFB signaling and ECM proteins involved in processes of extracellular structure and cellular adhesion (Figure 4B). In particular, THBS1, FN1, and COL1A2 appeared to be focal points connecting these processes (Figure 4B).
Bidirectional ECM/myocyte signaling in ICM. The bidirectional nature of ECM signaling with cells indicates that its composition likely plays a greater role in disease progression beyond deposition following injury and cardiomyocyte death. In ICM, the mechanical properties of ECM are altered (35–37), which may enhance pro-fibrotic TGFB signaling and further drive fibrosis progression. This concept is supported by studies showing that engineered heart tissues derived from stem cells grown on ECM from hypertrophic hearts exhibited contractile dysfunction (38). Consistent with these findings, our data reveal alterations in both the ECM and the contractile machinery of cardiomyocytes. Figure 5A shows a schematic of the connective proteins linking ECM to internal contractile machinery via costameres (39), and Figure 5B shows the significant proteins in our experiment corresponding to these linkages. Costameres are similar to the focal adhesions cardiomyocytes have at their distal ends but connect laterally to form sarcolemmal-cytoskeletal attachments (35, 39). In addition, costameric proteins shown to interact with integrins such as kindlins (FERMT2/3) and parvins also significantly change at end-stage ICM. FERMT2 (also known as kindlin-2) is an essential component of vertebrate intercalated discs and is essential for cytoskeletal organization and membrane attachment during development (40).
 Figure 5
Figure 5Bidirectional communication between ECM and cardiomyocytes contributes to ICM. (A) A general scheme for interaction between ECM and cardiomyocyte contractile machinery is shown. At the sarcolemma, integrins and other costameric proteins transmit forces from ECM to cytoskeleton and then to sarcomere. DGC, dystrophin glycoprotein complex. (B) Depiction of significantly altered protein groups from our data and the corresponding protein-protein interactions plotted in Cytoscape (93). (C) Heatmap and box plots of specific protein groups, demonstrating increased expression of ECM consistent with fibrosis and changes to intracellular components of cardiomyocytes, including fermitins, actinins, and myosin heavy-chain isoforms. For the box plots, ITGB2, FERMT3, MYH6, and MYH10 are plotted from the Azo extract, while PARVB, ITGB1BP2, TPM2, and MYOM2 are plotted from the LiCl extract. Box plots show the interquartile range, median (line), and minimum and maximum (whiskers). Significance testing was performed using limma. Then P values were adjusted by independent hypothesis weighting and plotted as follows: * ≤ 0.05, ** ≤ 0.01, *** ≤ 0.001.
We observed a significant downregulation of kindlin-2 in ICM, consistent with previous studies in mice showing that deletion of this gene results in HF and premature death (41). Interestingly, loss of FERMT2 was contrasted by upregulation of FERMT3 and ITGB2, both of which are exclusively expressed in hematopoietic cells. The interaction of FERMT3/ITGB2 is particularly important in platelet aggregation and innate immune response (42, 43) and is associated with unstable atherosclerotic plaques (44). We also observed the significant downregulation of muscle-specific integrin-binding protein melusin (ITGB1BP2), which protects the heart during ischemic injury via integrin-dependent activation of PI3K/AKT and ERK pathways (45). Figure 5C shows the specific proteins that changed at the interconnected stages of ECM, costamere/sarcolemma, and sarcomere. These include the adhesive glycoproteins like fibronectin, thrombospondins, and tenascins, which all increased in ICM, and components of the costamere like kindlins, parvins, and melusin. Intracellularly, myosin heavy chain 6 greatly decreased, as did tropomyosin-2 and myomesin-2, while myosin heavy chain 10 increased. Overall, there was a trend toward decreased expression of the intracellular components relative to the extracellular proteins. It is likely this is attributable at least in part to replacement fibrosis — cell death decreases the relative expression of intracellular components as ECM deposition takes the place of cells. Despite these contributions, interstitial fibrosis resulting in intracellular change is also very likely, given most patients in this study had no history of MI and had only experienced chronic ischemia.
 in failing ICM and nonfailing donor heart tissues.")







