Oncology
Abstract
Authors
Carolina M. Larrain, Jack H. Victory, Priyanka P. Desai, Lindsay R. Friedman, Hannah Stepp, Rachel Ashe, Kirsten Remmert, Surajit Sinha, Emily C. Smith, Nicole Russell, Tracey Pu, Alyssa V. Eade, Justine F. Burke, Jason Ho, Michael B. Yaffe, David E. Kleiner, Keith Schmidt, William D. Figg, Jonathan M. Hernandez
Abstract
BACKGROUND. Predictive biomarkers to guide chemotherapy decisions for metastatic castration resistant prostate cancer (mCRPC) are lacking. Preclinical studies indicate that circulating tumor cell (CTC) studies of chromosomal instability (CTC-CIN) can predict taxane resistance. METHODS. The CARD trial randomized subjects with mCRPC progressing within a year of treatment with an androgen receptor pathway inhibitor (ARPI; enzalutamide or abiraterone acetate plus prednisolone/prednisone) to cabazitaxel or the alternative ARPI. As a pre-planned biomarker analysis, CTCs were isolated from blood samples obtained at baseline; cycle two, and end of treatment. Associations between baseline CTC and CTC-CIN counts with imaging-based progression free survival (ibPFS), overall survival (OS), time to prostate-specific antigen (PSA) progression, RECIST 1.1 objective response rate (ORR), and PSA50 response rate (PRR) were assessed. RESULTS. High baseline CTC-CIN counts significantly associated with worse OS after adjustment for confounding variables (median OS, 15.3 vs 8.9 months; univariate HR, 2.16; 95% CI, 1.52 – 3.06; p < 0.001; multivariate HR, 1.56; 95% CI, 1.01 – 2.43; p = 0.047). Detectable CTC-CIN counts at baseline may predict a lack of ibPFS and OS benefit when comparing cabazitaxel to ARPI. CONCLUSION. This preplanned biomarker analysis of CARD confirms that CTC-CIN counts are a clinically useful prognostic and predictive biomarker of taxane resistance in mCRPC. Detectable CTC-CIN at baseline defines a patient subpopulation with unmet clinical needs in which alternative therapeutics should be tested. TRIAL REGISTRATION. CARD ClinicalTrials.gov number, NCT02485691. FUNDING. Funded by Sanofi and Epic Sciences.
Authors
Ossian Longoria, Jan Rekowski, Santosh Gupta, Nick Beije, Klaus Pantel, Eleni Efstathiou, Cora Sternberg, Daniel Castellano, Karim Fizazi, Bertrand Tombal, Adam Sharp, Oliver Sartor, Sandrine Macé, Christine Geffriaud-Ricouard, Richard Wenstrup, Ronald de Wit, Johann de Bono

Abstract
Macrophage migration inhibitory factor (MIF) is an upstream regulatory cytokine that is associated with advanced disease and poor outcomes in multiple cancer types, including melanoma. We investigated whether anti-MIF therapy could enhance the antitumor effects of the immune checkpoint inhibitor anti–programmed cell death 1 (anti–PD-1) in 2 murine tumor models. The therapeutic efficacy of anti-MIF, alone or combined with anti–PD-1, was tested in the YUMMER1.7 melanoma and MC38 colorectal cancer models. Tumor growth and survival were assessed in untreated Mif-knockout (KO) and low-expression human MIF allele (CATT5) mice and compared with wild-type (WT) or high-expression MIF allele (CATT7) mice. Tumor-bearing animals underwent cytokine profiling, tumor immunohistochemistry, flow cytometry, and scRNA-Seq. We also correlated functional variant MIF alleles with melanoma incidence and progression in patients. Our results showed that combined anti-MIF and anti–PD-1 significantly reduced tumor growth, improved survival, and promoted tumor regression, accompanied by enhanced TH1 cytokine levels, increased macrophage activation–related cytokines, and increased type 1 conventional dendritic cells. scRNA-Seq analysis revealed an expansion of intratumor Cd74/C1q/Aif1-expressing macrophages, which exhibited an antitumor phenotype, in response to anti-MIF therapy. MIF-KO and CATT5 mice exhibited reduced tumor burdens compared with WT or CATT7 mice alone and in the presence of anti–PD-1. In patients with melanoma, the high-MIF expression genotype (-173C/C) occurred at higher frequencies compared with healthy controls. These findings highlight that the addition of anti-MIF to anti–PD-1 reduces tumor growth, enhances antitumor responses, prolongs survival, and augments key intratumor immune cell populations involved in immune activation against tumors. This approach merits further consideration for clinical trial development.
Authors
Thuy T. Tran, Gabriela Athziri Sánchez-Zuno, Lais Osmani, Jasmine Caulfield, Caroline Naomi Valdez, Marta Piecychna, Lin Leng, Michelle E. Armstrong, Seamas C. Donnelly, Carlo B. Bifulco, Terri Clister, Rajan P. Kulkarni, Lin Zhang, Mario Sznol, Lucia Jilaveanu, Harriet M. Kluger, Insoo Kang, Richard Bucala
Abstract
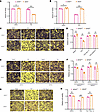
Apoptosis and necroptosis are 2 distinct destinies of cells stimulated with TNF-α; however, it remains unclear how apoptosis and necroptosis are differentially regulated. This study validates the key regulatory role of speckle-type POZ protein (SPOP) in balancing apoptosis and necroptosis. SPOP promotes the polyubiquitination and degradation of receptor-interacting serine/threonine-protein kinase 3 (RIPK3), thereby inhibiting necrosome formation and decreasing cellular sensitivity to necroptosis. Conversely, SPOP interacted with RIPK1 independently of its E3 ubiquitin ligase activity, protecting it from ubiquitination and degradation, thereby enhancing RIPK1 expression and cellular sensitivity to apoptosis. Inhibiting RIPK1 kinase activity with 7-Cl-O-Nec-1 impeded both SPOP-mediated apoptosis and SPOP deficiency–mediated necroptosis. Besides, inhibition or loss of RIPK3 rescued SPOP deficiency–mediated necroptosis. Pancancer analyses indicated that the SPOP/RIPK1/RIPK3 axis is dysfunctional in a variety of tumors. In 3 representative tumor types with high expression of SPOP and RIPK1, kidney renal clear cell carcinoma, liver hepatocellular carcinoma, and breast invasive carcinoma, this regulatory mechanism remains applicable. Based on these findings, a combination therapy using the second mitochondria-derived activator of caspases (Smac) mimetic SM164 and sunitinib was developed, demonstrating a more pronounced efficacy than sunitinib monotherapy, and this sensitizing effect was dependent on the expression level of RIPK1. These results suggest that the combination of Smac mimetics with tyrosine kinase inhibitors holds potential clinical value for tumors with dysregulated SPOP/RIPK1/RIPK3 signaling.
Authors
Yuzhong Ye, Changjie Yue, Zaosong Zheng, Hailong Ruan, Yuanpeng Zhang, Qi Miao, Xiaoping Zhang, Wen Xiao, Lei Liu
Abstract
BACKGROUND Cisplatin is often the cytotoxic drug of choice for chemoradiation therapy (CRT) for head and neck squamous cell carcinoma (HNSCC), but it can lead to irreversible hearing loss. There may be similar oncologic outcomes but different toxicity profiles depending on whether cisplatin is given at 75–100 mg/m2 every 3 weeks or 30–40 mg/mg2 weekly. This study compares cisplatin-induced hearing loss in patients with HNSCC receiving similar cumulative doses of cisplatin administered either on higher-dose or lower-dose treatment schedules.METHODS Using the Enhancing Cancer Hearing Outcomes (ECHO) dataset from 5 academic centers, we conducted a multicenter retrospective cohort study of adults (≥18 years) with HNSCC receiving cisplatin-based CRT. Participants were grouped by cisplatin dose schedule: every 3 weeks (≥75 mg/m²) or weekly (<75 mg/m²). Hearing loss was assessed using American Speech-Language-Hearing Association (ASHA) and Common Terminology Criteria for Adverse Events (CTCAE) v5.0 threshold shift criteria based on audiograms obtained ≤120 days before and after treatment. Risk differences and predictors of hearing loss were evaluated using χ2 analyses and multivariate regression. Kaplan-Meier curves assessed overall and disease-free survival.RESULTS Among 564 participants (1,127 ears), lower-dose weekly cisplatin was associated with significantly lower incidence of hearing loss (ASHA criteria: 57% vs. 82%; CTCAE criteria: 39% vs. 69%). CTCAE grade ≥2 hearing loss occurred in 18% of the weekly group versus 50% of the 3-week group. Multivariate analysis confirmed treatment schedule as an independent predictor of ototoxicity. Two-year survival outcomes did not differ between groups.CONCLUSIONS Weekly low-dose cisplatin significantly reduced the incidence and severity of hearing loss without compromising survival, supporting its broader use in CRT for HNSCC.
Authors
Katharine A. Fernandez, Abu S. Chowdhury, Amanda Bonczkowski, Paul D. Allen, Maura H. Campbell, David S. Lee, Charvi Malhotra, Brandi R. Page, Deborah A. Mulford, Candice Evita Ortiz, Peter L. Santa Maria, Peter Kullar, Saad A. Khan, Shawn D. Newlands, Nicole C. Schmitt, Lisa L. Cunningham
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a rapidly metastasizing cancer characterized by a dense desmoplastic stroma comprised of extracellular matrix (ECM) proteins, which complicates treatment. Upon stimulation, pancreatic stellate cells (PSCs) differentiated into cancer-associated fibroblasts (CAFs) that are the source of ECM and cytokines in PDAC. We previously reported that mechanical stress activates PSCs and induces fibrosis through mechanical ion channel PIEZO1-mediated TRPV4 channel activation, but its role in PDAC remains unclear. Here we report that pathological activation of PIEZO1 differentiated human PSCs into an inflammatory CAF phenotype that expresses chemoresistance and cancer stemness markers CD10 and GPR77. In an orthotopic PDAC model, TRPV4 knockout mice exhibited a significant reduction in tumor size, circulating inflammatory cytokines, tissue inhibitor of metalloproteinases-1 (TIMP1), and pre-metastatic niche markers, serum amyloid A (SAA) proteins. A similar trend was observed in mice lacking functional PIEZO1 in PSCs. The livers of TRPV4 knockout mice exhibited fewer cancer cell microlesions, lacked macro tumors, produced lower levels of inflammatory protein S100A8, and developed fewer inflammatory cell clusters. In orthotopic and genetically engineered models of PDAC, these mice also had improved survival, suggesting that blocking TRPV4 channels may be a promising therapeutic target for PDAC.
Authors
Joelle M.-J. Romac, Sandip M. Swain, Nidula Mullappilly, Bandana Bindhani, Rodger A. Liddle
Abstract
IDH1/2 mutations (IDHmut) increase methylation of DNA and histones in gliomas. IDHmut inhibitors are effective against low-grade IDHmut gliomas, but new strategies against high grade IDHmut gliomas are needed. Although histone deacetylase inhibitors (HDACi) are ineffective against IDHwt glioblastoma (GBM), their potential in IDHmut gliomas has not been extensively studied. We previously established that IDHmut gliomas are more sensitive to HDACi than IDHwt GBM. Here we show that IDHmut is associated with greater sensitivity to HDACi only in glioma, not in IDHmut chondrosarcoma or cholangiocarcinoma. While HDACi induced more histone acetylation and gene regulation in IDHmut glioma than in IDHwt GBM, such acetylation was mostly within gene deserts, whereas IDHmut glioma promoters paradoxically lost histone acetylation. Two mediators of HDACi resistance, YAP and TAZ, were methylated and suppressed in IDHmut gliomas, but not in other IDHmut cancers. Inducing YAP or TAZ expression in IDHmut gliomas conferred resistance to HDACi. Finally, belinostat extended in vivo survival only in IDHmut glioma models, not in IDHmut GBM models. Our findings provide a mechanistic rationale for further studies of HDACi in IDHmut glioma patients, as well as the potential use of YAP/TAZ as a biomarker of HDACi sensitivity in cancers.
Authors
Thomas K. Sears, Matthew McCord, Wenxia Wang, Alicia Steffens, Kathleen McCortney, Rahul Chaliparambil, Jann N. Sarkaria, Craig M. Horbinski

Abstract
Epigenetic scarring of terminally dysfunctional (TDysf) CD8+ T cells hinders long-term protection and response to immune checkpoint blockade during chronic infections and cancer. We developed a faithful in vitro model for CD8+ T cell terminal dysfunction as a platform to advance T cell immunotherapy. Using TCR-transgenic CD8+ T cells, we found that 1-week peptide stimulation, mimicking conditions in previous models, failed to induce a stable exhaustion program. In contrast, prolonged stimulation for 2–3 weeks induced T cell dysfunction but triggered activation-induced cell death, precluding long-term investigation of exhaustion programs. To better mimic in vivo exhaustion, we provided post-effector, chronic TGF-β1 signals, enabling survival of chronically stimulated CD8+ T cells for over 3 weeks. These conditions induced a state of terminal dysfunction, marked by a stable loss of effector, cytotoxicity, and memory programs, along with mitochondrial stress and impaired protein translation. Importantly, transcriptomic and epigenetic analyses verified the development of terminal exhaustion-specific signatures in TDysf cells. Adoptive transfer of TDysf cells revealed their inability to recall effector functions or proliferate after acute lymphocytic choriomeningitis virus rechallenge. This tractable model system enables investigation of molecular pathways driving T cell terminal dysfunction and discovery of therapeutic targets for cancer or chronic infections.
Authors
Amir Yousif, Abbey A. Saadey, Ava Lowin, Asmaa M. Yousif, Ankita Saini, Madeline R. Allison, Kelley Ptak, Eugene M. Oltz, Hazem E. Ghoneim
Abstract
BACKGROUND. Cardiotoxicity is a major complication of anti-cancer therapy (CTx); yet, the impact of CTx on the human microcirculation is not well defined. This study evaluated the impact of CTx on microvascular function in breast cancer patients. METHODS. Endothelial function and angiogenic potential were assessed in arterioles and adipose biopsies obtained from breast cancer patients before, during, and after CTx (longitudinal and cross-sectional) and in healthy arterioles exposed to doxorubicin (Dox), trastuzumab (TZM), or paclitaxel (PTX) ex vivo. Conditioned media containing VEGF-B protein was used to test feasibility of a targeted intervention. RESULTS. Patients treated with Dox and/or TZM in vivo developed profound microvascular endothelial dysfunction that persisted for ≥9 months after treatment cessation. Angiogenic potential was reduced during CTx and recovered within one month after cessation. Gene expression related to angiogenesis and inflammation changed over the course of clinical treatment. Isolated adipose arterioles from healthy donors developed endothelial dysfunction when exposed to Dox or TZM ex vivo. In contrast, paclitaxel (PTX), which poses minimal cardiovascular risk, had no impact on vasomotor function. Ex vivo exposure to Dox or PTX suppressed angiogenic potential, whereas TZM had no effect. Treatment with VEGF-B protein preserved endothelial function in healthy arterioles exposed to Dox or TZM ex vivo. CONCLUSION. Breast cancer patients undergoing treatment with Dox and/or TZM develop prolonged microvascular endothelial dysfunction that is recapitulated in healthy arterioles exposed to Dox or TZM ex vivo. Targeted intervention with VEGF-B protects against direct Dox- or TZM-induced vascular toxicity in human arterioles ex vivo. FUNDING. National Institutes of Health grant R01 HL133029, HL173549 (AMB). National Institutes of Health grant T32 HL134643 (JDT, STH). American Heart Association grant SFRN847970 (AMB, DDG). We Care Foundation Grant (AMB, ALK). Medical College of Wisconsin Cardiovascular Center Pre-PPG Grant (AMB). Advancing a Healthier Wisconsin – Redox Biology Grant (AMB). Jenny and Antti Wihuri Foundation (RMK).
Authors
Janée D. Terwoord, Laura E. Norwood Toro, Shelby N. Hader, Stephen T. Hammond, Joseph C. Hockenberry, Jasmine Linn, Ibrahim Y. Vazirabad, Amanda L. Kong, Alison J. Kriegel, Ziqing Liu, Riikka M. Kivelä, Gillian Murtagh, David D. Gutterman, Andreas M. Beyer
Abstract
Glioblastoma IDH-wildtype is the most common and aggressive primary brain tumor in adults, with poor prognosis despite current therapies. To identify new therapeutic vulnerabilities, we investigated the role of CDK12, a transcription-associated cyclin-dependent kinase, in glioblastoma. Genetic or pharmacologic inactivation of CDK12 impaired tumor growth in patientderived xenograft (PDX) models and enhanced the efficacy of temozolomide. Metabolic profiling using extracellular flux analysis and stable isotope tracing with U-¹³C-glucose and U-¹³Cglutamine showed that CDK12 inhibition disrupted mitochondrial respiration, resulting in energy depletion and apoptotic cell death characterized by caspase activation and Noxa induction. Mechanistically, we identified a direct interaction between CDK12 and GSK3β. CDK12 inhibition activated GSK3β, leading to downregulation of PPARD, a transcriptional regulator of oxidative metabolism. This CDK12–GSK3β–PPARD axis was required for glioblastoma cell proliferation and metabolic homeostasis. In vivo, CDK12 inhibition significantly extended survival without overt toxicity and induced complete tumor regression in a subset of animals. Strikingly, combined CDK12 inhibition and temozolomide treatment led to complete tumor eradication in all animals tested. These findings establish CDK12 as a key regulator of glioblastoma metabolism and survival, and provide strong preclinical rationale for its therapeutic targeting in combination with standard-of-care treatments.
Authors
Jeong-Yeon Mun, Chang Shu, Qiuqiang Gao, Zhe Zhu, Hasan O. Akman, Mike-Andrew Westhoff, Georg Karpel-Massler, Markus D. Siegelin
No posts were found with this tag.


