Immunology
Abstract
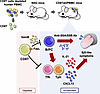
Cytomegalovirus (CMV) is a prevalent β-herpesvirus that persists asymptomatically in immunocompetent hosts. In people with HIV-1 (PWH), CMV is associated with HIV-1 persistence and particular inflammatory-related co-morbidities. The true causative role of CMV in HIV-associated pathologies however remains unclear given that nearly all PWH are coinfected with CMV. In this study, we examined acute phase immune and virological dynamics in cohorts of SIV-infected rhesus macaques (RMs) that were naturally seropositive or -negative for rhesus CMV (RhCMV). We observed prior to SIV, RhCMV expanded a polyclonal population of target CCR5+CD4+ T cells in gut and lymph nodes (LN) that expressed the chemotactic receptor CXCR3 and were largely not specific for RhCMV. Upon SIV infection, RhCMV+ RMs exhibited higher peak viremia and elevated levels of SIV DNA in the upper and lower intestine. Greater seeding of SIV DNA was associated with a maintenance of CCR5-expressing CD4+ T cells that were enriched within the RhCMV+ gut along a CXCR3-CXCL9 chemotactic axis. Overall, the data suggest that RhCMV can promote SIV susceptibility within a diverse, polyclonal pool of CD4 T cells that are not entirely RhCMV-specific.
Authors
Chrysostomos Perdios, Naveen Suresh Babu, Celeste D. Coleman, Anna T. Brown, Shevon N. Alexander, Matilda J. Moström, Carolina Allers, Lara Doyle-Meyers, Christine M. Fennessey, Lori A. Rowe, Brandon F. Keele, Amitinder Kaur, Michael L. Freeman, Joseph C. Mudd
Abstract
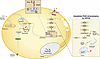
Peripheral helper T (Tph) and follicular helper T (Tfh) cells are key regulators of B cell differentiation and antibody production, making them promising targets for autoimmune disease treatment. However, their differentiation mechanisms differ significantly between humans and mice, limiting drug validation in mouse models. Here, we present a simple and effective method for in vivo proliferation of human Tph/Tfh and B cells. We discovered that after depleting CD8+ T cells of human peripheral blood mononuclear cell–transferred immunodeficient mice (CD8TΔhPBMC mice), human Tph/Tfh cells and B cells proliferated markedly in the spleen compared with those in human PBMC–transferred immunodeficient mice (hPBMC mice). Transcriptome analysis confirmed proliferating cells’ close resemblance to human Tph/Tfh cells. Furthermore, multicolor flow cytometry revealed CXCL13+ Tph cells infiltrating Sjögren’s syndrome–associated (SjS-associated) organs, such as salivary glands. Single-cell RNA sequencing identified IL-21+CXCL13+IFN-γ+ICOS+TIGIT+GPR56+ Tph cells in the salivary glands. These findings are consistent with reduced saliva volume and elevated SjS markers, such as anti-SSA antibody, in these mice, which were both ameliorated by immunosuppressants. In vitro, CD8+ T cells from hPBMC mice induced B cell apoptosis and inhibited Tph/Tfh differentiation. This model advances understanding of human Tph/Tfh cell biology and offers a valuable platform for studying SjS and therapeutic targets.
Authors
Mariam Piruzyan, Sota Fujimori, Ryota Sato, Yuki Imura, Sachiko Mochiduki, Kana Takemoto, Akiko Nishidate, Yuzo Koda
Abstract
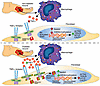
Processes that promote white adipocyte inflammatory function remain incompletely defined. Here, we demonstrated that type I interferon–dependent (IFN-I–dependent) skewing of adipocyte glycolysis, nicotinamide adenine dinucleotide (NAD+) utilization, and pyruvate kinase isozyme M2 (PKM2) function may contribute to increased systemic and tissue inflammation and disease severity in obesity. Notably, chemical and/or genetic inhibition of glycolysis, the NAD+ salvage pathway, or PKM2 restricted IFN-I–dependent increase in adipocyte inflammatory cytokine production. Further, genetic or small molecule targeting of PKM2 function in vivo was sufficient to reduce systemic and tissue inflammation and metabolic disease severity in obese mice, in an adipocyte PKM2-dependent manner. Further, white adipose tissue of individuals living with obesity and metabolic disease, compared with metabolically healthy individuals with obesity, showed an increase in expression of inflammatory and metabolic genes, while small molecule targeting of PKM2 function contributed to reduced IFN-I–driven inflammatory cytokine production by primary human adipocytes. Together, our findings invoke the IFN-I/PKM2 axis as a potential target for modulating adipocyte dysregulated inflammation.
Authors
Michelle S.M.A. Damen, Pablo C. Alarcon, Calvin C. Chan, Traci E. Stankiewicz, Hak Chung, Keisuke Sawada, Cassidy J. Ulanowicz, John Eom, Jarren R. Oates, Jennifer L. Wayland, Jessica R. Doll, Rajib Mukherjee, Miki Watanabe-Chailland, Lindsey Romick-Rosendale, Sara Szabo, Michael A. Helmrath, Joan Sanchez-Gurmaches, Maria E. Moreno-Fernandez, Senad Divanovic
Abstract
Fibroblast to myofibroblast transition is a critical event required for effective tissue repair. In pathologic wound repair processes, such as type 2 diabetes (T2D), fibroblast to myofibroblast transition is impaired. The exact factors that control this transition in wounds are unclear. Here, using human tissue and murine transgenic models, we show that the histone methyltransferase SETDB2 is elevated in diabetic wound fibroblasts and TNF-α represses fibroblast to myofibroblast transition via Setdb2. We identified that TNF-α increases Setdb2 in fibroblasts via a JAK1,3/STAT3 signaling pathway, where pharmacologic or genetic manipulation of this pathway altered Setdb2 in fibroblasts. We also found that fibroblasts treated with pro-inflammatory macrophage supernatants displayed increased Setdb2 and downregulated myofibroblast genes; inhibition of the TNF-α receptor reduced the upregulation of Setdb2. In diabetes, we showed that TNF-α signaling was increased in wound fibroblasts, which functions to increase Setdb2 expression and represses fibroblast to myofibroblast transition. Fibroblast-specific knockdown of SETDB2 and therapeutic inhibition of JAK1,3/STAT3 improved diabetic wound repair, where wound fibroblasts expressed increased myofibroblast genes. This study is the first to our knowledge to identify an epigenetic mechanism for reduced fibroblast to myofibroblast transition in diabetic wounds. Therapeutic targeting of the TNF-α/STAT3/SETDB2 axis in wound fibroblasts may improve diabetic wound healing.
Authors
Tyler M. Bauer, Kevin D. Mangum, Samuel D. Buckley, James Shadiow, Amrita D. Joshi, Christopher O. Audu, Jadie Y. Moon, Lindsey D. Hughes, Rachel Bogel, Lam C. Tsoi, Qinmennge Li, He Zhang, Steven Kunkel, Johann E. Gudjonsson, Frank M. Davis, Katherine A. Gallagher
Abstract
Juvenile idiopathic arthritis (JIA) is the most prevalent chronic inflammatory arthritis of childhood, yet the spatial organization in the synovium remains poorly understood. Here, we perform subcellular-resolution spatial transcriptomic profiling of synovial tissue from patients with active JIA. We identify diverse immune and stromal cell populations and reconstruct spatially defined cellular niches. Applying a newly developed spatial colocalization analysis pipeline, we uncover microanatomical structures, including endothelial–fibroblast interactions mediated by NOTCH signalling, and a CXCL9-CXCR3 signaling axis between inflammatory macrophages and CD8+ T cells, alongside the characterization of other resident macrophage subsets. We also detect and characterize tertiary lymphoid structures marked by CXCL13-CXCR5 and CCL19-mediated signaling from Tph cells and immunoregulatory dendritic cells, analogous to those observed in other autoimmune diseases. Finally, comparative analysis with rheumatoid arthritis reveals JIA-enriched cell states, including NOTCH3+ and CXCL12+ sublining fibroblasts, suggesting potentially differential inflammatory programs in pediatric versus adult arthritis. These findings provide a spatially resolved molecular framework of JIA synovitis and introduce a generalizable computational pipeline for spatial colocalization analysis in tissue inflammation.
Authors
Jun Inamo, Roselyn Fierkens, Michael R. Clay, Anna Helena Jonsson, Clara Lin, Kari Hayes, Nathan D. Rogers, Heather Leach, Kentaro Yomogida
Abstract
Authors
Zhehao Tan, Gio Wu, Daniela Salgado Figueroa, Paramita Dutta, Zachary Jaeger, Marissa Mazurie, David Schairer, Dawn Eichenfield, Wynnis L. Tom, Lauren Galli, Lawrence Eichenfield, Bob Geng, Brian Hinds, Hal M. Hoffman, Lori Broderick, Ben Croker, Ferhat Ay, Reid Oldenburg
Abstract
BACKGROUND. Cannabidiol (CBD) is increasingly used for pain management, including in transplant recipients with limited analgesic options. Its immunomodulatory effects in humans are not well defined at a single cell level at CBD steady state with concomitant tacrolimus treatment. METHODS. In a Phase 1 ex vivo study, peripheral blood mononuclear cells from 23 participants who received oral CBD (Epidiolex®) up to 5 mg/kg twice daily for 11 days were collected before CBD (pre-CBD) and at steady state (post-CBD). Lymphocytes were isolated and stimulated with anti-CD3/CD28 antibodies, with or without tacrolimus (5 ng/mL). Pharmacodynamic responses were assessed using CellTiter-Glo® proliferation, single-cell/nucleus RNA sequencing, cytokine assays, and flow cytometry. Steady-state plasma concentrations of CBD were quantified via tandem mass spectrometry. RESULTS. We identified an increased proportion of T effector memory (TEM) cells post-cannabidiol (22% increase), which correlated with CBD plasma concentrations (R = 0.77, P-value = 0.01). Cannabidiol reduced proliferation of T (37% decrease) and CD70hi B (17% decrease) lymphocytes with additive immunosuppressive effects to tacrolimus. Single-cell RNA sequencing revealed reduced IL2 and TNF signaling and altered receptor–ligand networks in TEM cells. Post-cannabidiol cytokine assays revealed elevated proinflammatory IL-6 protein levels and anti-inflammatory IL-10 levels, with reduced TNF-α, LTA, and IL-2. In flow cytometry, the proportion of TEM and TEMRA increased post-cannabidiol with tacrolimus. CONCLUSION. Cannabidiol exhibits mixed immunomodulatory effects with pro- and anti-inflammatory signals. Understanding the clinical safety of cannabidiol use is important given the paucity of pain control options available for immunocompromised transplant populations.
Authors
Debora L. Gisch, Sachiko Koyama, Jumar Etkins, Gerald C. So, Daniel J. Fehrenbach, Jessica Bo Li Lu, Ying-Hua Cheng, Ricardo Melo Ferreira, Evan Rajadhyaksha, Kelsey McClara, Mahla Asghari, Asif A. Sharfuddin, Pierre C. Dagher, Laura M. Snell, Meena S. Madhur, Rafael B. Polidoro, Zeruesenay Desta, Michael T. Eadon
Abstract
BACKGROUND. Fecal Microbiota Transplantation (FMT) is the most effective therapy for recurrent Clostridioides difficile infection (rCDI), yet its mechanism of action remains poorly understood. METHODS. We report the results of a clinical trial of subjects undergoing FMT therapy for rCDI (n=16), analyzing colon biopsies, plasma, peripheral blood mononuclear cells, and stool at the time of FMT and two-month follow-up. Plasma and colon biopsy samples were also collected from healthy controls for comparison with rCDI patients. Microbiome composition, colonic gene expression, and immune changes were evaluated through high-throughput sequencing and immunoprofiling via flow cytometry. RESULTS. No subjects experienced recurrence at follow-up. FMT significantly altered the intestinal microbiome but had no significant impact on the systemic immune system. In contrast, FMT promoted broad changes in colonic transcriptional profiles compared to both pre-FMT and healthy control biopsies, inhibiting genes associated with pro-inflammatory signaling and upregulating type 2 immunity and proliferative pathways (Myc and mTORC1). FMT increased expression of IL-33 and the type 2 immune EGFR family ligand amphiregulin, potentially explaining upregulation of Myc and mTORC1 pathways. Spatial transcriptomics demonstrated that these changes were localized to the colonic epithelium. Comparison of transcriptional profiles with available single cell gene sets determined that post-FMT biopsies were enriched in signatures associated with proliferative cell types while repressing signatures of differentiated colonocytes. CONCLUSIONS. We conclude that FMT promotes proliferation of the colonic epithelium in rCDI patients, which may drive regeneration and protect against subsequent CDI. REGISTRATION. Clinicaltrials.gov NCT02797288. FUNDING. NIH grants R01 AI152477, R01 AI124214, and K23 AI163368.
Authors
G. Brett Moreau, Jiayi Tian, Nick R. Natale, Farha Naz, Mary K. Young, Uma Nayak, Mehmet Tanyüksel, Isaura Rigo, Gregory R. Madden, Mayuresh M. Abhyankar, Nicholas Hagspiel, Savannah Brovero, Mark Worthington, Brian Behm, Chelsea Marie, William A. Petri Jr., Girija Ramakrishnan
Abstract
Herpes Simplex Virus 2 (HSV-2) infection results in variable rates of local viral shedding in anogenital skin. The impact of episodic viral exposures on immune cells in adjacent mucosal tissues, including the genital tract, is unknown. However, any immune responses at this site could impact protective mucosal immunity, tissue homeostasis, and adverse health outcomes. To investigate the impact of HSV-2 on cervicovaginal tract immunity, we applied flow cytometry, immunofluorescent imaging, analysis of soluble immune factors, and spatial transcriptomics to cervicovaginal tissue and blood samples provided by a total of 232 HSV-2-seropositive and seronegative participants, with genital HSV-2 shedding evaluated at the time of biopsy. This unique dataset was used to define and spatially map immune cell subsets and localized gene expression via spatial transcriptomics. HSV-2-seropositivity alone was associated with minimal differences in cervicovaginal and circulating T cell phenotypes. However, the vaginal mucosa during active HSV-2 shedding was associated with alterations in T cell, macrophage, and dendritic cell localization and gene expression consistent with increased immune surveillance, with immune activating and suppressing signals potentially reinforcing mucosal tissue homeostasis.
Authors
Finn MacLean, Rachael M. Zemek, Adino Tesfahun Tsegaye, Jessica B. Graham, Jessica L. Swarts, Sarah C. Vick, Nicole B. Potchen, Irene Cruz Talavera, Lakshmi Warrier, Julien Dubrulle, Lena K. Schroeder, Anna Elz, David Sowerby, Ayumi Saito, Katherine K. Thomas, Matthias Mack, Joshua T. Schiffer, R. Scott McClelland, Keith R. Jerome, Bhavna H. Chohan, Kenneth Ngure, Nelly Rwamba Mugo, Evan W. Newell, Jairam R. Lingappa, Jennifer M. Lund
Abstract
Patients with cutaneous T cell lymphoma (CTCL) experience high morbidity and mortality due to S. aureus skin infections and sepsis, but the underlying mechanisms remain unclear. We have previously identified high levels of LAIR2, a decoy protein for the inhibitory receptor LAIR1, in advanced CTCL. Mice lack a LAIR2 homolog, so we used Lair1 knock-out (KO) mice to model LAIR2 overexpression. In a model of S. aureus skin infection, Lair1 KO mice had significantly larger abscesses and areas of dermonecrosis compared to WT despite similar bacterial burdens. Lair1 KO exhibited a pattern of increased inflammatory responses in infection and sterile immune stimulation, with increased production of proinflammatory cytokines and myeloid chemokines, neutrophil ROS, and collagen/ECM pathway proteins, including collagens and complement factors. These findings support the notion that loss of LAIR1 signaling causes an excessive inflammatory response that exacerbates tissue damage and does not improve infection control. Underscoring the clinical relevance of our findings, CTCL skin lesions exhibited similarly increased expression in cytokine and collagen/ECM remodeling pathways, suggesting that high levels of LAIR2 promote excessive inflammatory tissue damage and compromise host defense against S. aureus infection. LAIR signaling represents a promising target for therapeutic development in CTCL and other inflammatory diseases.
Authors
Hannah K. Dorando, Evan C. Mutic, Kelly L. Tomaszewski, Yulia Korshunova, Ling Tian, Mellisa K. Stefanov, Chaz C. Quinn, Deborah J. Veis, Juliane Bubeck Wardenburg, Amy C. Musiek, Neha Mehta-Shah, Jacqueline E. Payton
No posts were found with this tag.


