Research ArticleCell biologyMuscle biology
Open Access | ![]() 10.1172/jci.insight.194581
10.1172/jci.insight.194581
Spatial proteomics reveals recombinant human laminin-111 restores adhesion signaling to laminin-α2–deficient muscle
Hailey J. Hermann,1 Ryan D. Wuebbles,1 Marisela Dagda,1 Axel Muñoz,1 Lauren L. Parker,1 Paula C. Guzman,1 Lola T. Byrne,1 Steven A. Moore,2 and Dean J. Burkin1
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Hermann, H. in: PubMed | Google Scholar
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Wuebbles, R. in: PubMed | Google Scholar
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Dagda, M. in: PubMed | Google Scholar
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Muñoz, A. in: PubMed | Google Scholar
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Parker, L. in: PubMed | Google Scholar
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Guzman, P. in: PubMed | Google Scholar
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Byrne, L. in: PubMed | Google Scholar
1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by
Moore, S.
in:
PubMed
|
Google Scholar
|

1Department of Pharmacology, Reno School of Medicine, University of Nevada, Reno, Nevada, USA.
2Department of Pathology, Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Dean J. Burkin, 1664 N. Virginia Street MS 573, Reno, Nevada 89557, USA. Phone: 775.784.6288; Email: dburkin@med.unr.edu.
Find articles by Burkin, D. in: PubMed | Google Scholar
Published October 21, 2025 - More info
JCI Insight. 2025;10(22):e194581. https://doi.org/10.1172/jci.insight.194581.
© 2025 Hermann et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: April 15, 2025; Accepted: October 2, 2025

-
Results
DSP analysis of patients with LAMA2-CMD and controls. Twelve individuals were included in this study: 6 patients with LAMA2-CMD and 6 age-matched unaffected controls. The University of Iowa collected all patient muscles under their IRB-approved protocol and provided specimens for research as deidentified muscle sections on glass slides. All biopsies were collected for initial diagnostic purposes. Due to limited availability, control tissue was age matched as closely as possible, and immunofluorescence (IF) was used to confirm the absence of laminin-α2 (Figure 1, A and B). Four out of 6 of the patients exhibited full laminin-211 deficiency (Figure 1, C and D), and 2 patients experienced partial laminin-α2 loss (Figure 1, E–H). Patient demographics and LAMA2 variants are listed in Table 1. Two different types of ROIs were selected from each patient tissue biopsy: a small representative area and a single skeletal muscle fiber that includes muscle fibers and the surrounding microenvironment (Figure 2, A and B). In patient samples, area ROIs were selected from regions showing minimal muscle damage and immune cell infiltration based on morphological markers for desmin, CD68, CD31, and DNA (Figure 2A). To investigate potential muscle-specific protein changes, single-fiber ROIs were also included in this study (Figure 2B). Hypertrophic fibers and low-desmin-expressing fibers were avoided. Using NanoString’s GeoMx IO Proteome Atlas (IPA), 575 different proteins were analyzed (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.194581DS1). After normalizing protein probe counts to negative controls (rabbit IgG, rat IgG2a, and rat IgG2b), heatmaps were generated through the Nanostring Data Analysis suite. Further downstream analysis in which volcano plots were generated identified differentially expressed proteins (DEPs), displaying 216 DEPs in the area ROIs and 40 DEPs in the single-fiber ROIs from the GeoMx IPA (Figure 2, C and D, and Supplemental Table 2). DEP analysis of the area ROIs revealed that out of the 216 DEPs, only 2 proteins were enriched in the unaffected group, and 214 were enriched in the patients with LAMA2-CMD (Figure 2E). The single-fiber ROI volcano plots displayed only 1 upregulated DEP in the unaffected group compared with 39 proteins in the LAMA2-CMD muscle fibers (Figure 2F). LAMA2-CMD DEPs from both ROI groups were then categorized using NanoString’s target group classification from the data analysis suite (Figure 2, G and H). Similar biological processes were observed in both area and single-fiber ROI data sets, including increased expression of proteins involved in the MAPK and PI3K/AKT pathways. Notably, the epithelial-mesenchymal transition (EMT) pathway had the highest number of DEPs in the single-fiber group. EMT results from transcription factors that alter gene expression to promote loss of cell-cell adhesion (19), suggesting a shift in cytoskeletal dynamics within the single fibers of patients with LAMA2-CMD (Figure 2H). Other protein biological process groups were associated with senescence, oxidative stress, and apoptosis. Furthermore, there was a large presence of DEPs associated with immune response activation, such as chemokine, interleukin, and TLR signaling (Figure 2, G and H).
 Figure 1
Figure 1Histopathology and laminin-211 IF of human muscle biopsies. (A) H&E-stained cryosections are shown for normal skeletal muscle, and (B) laminin-211 IF stained cryosections of normal skeletal muscle. (C) H&E-stained cryosections for severe LAMA2-CMD (case 1) and (D) laminin-stained IF. (E) Mild, partially LAMA2-CMD (case 4) histopathology and (F–H) laminin-211 IF staining using antibodies against C-terminus, middle portion, and N-terminus of laminin-211. Scale bars: 50 μm (A, C, and D) and 100 μm (B and E–H).
 Figure 2
Figure 2Spatial profiling reveals large-scale changes in LAMA2-CMD patient muscle. (A) Selected area ROIs from unaffected and patient tissues based on morphological markers DNA, desmin, CD31, and CD68. Scale bar: 500 μm. (B) Single-fiber ROIs from unaffected and patient tissues were selected based on desmin expression. Scale bar: 50 μm. (C and D) Protein expression heatmaps of significantly differentially expressed proteins (DEPs) from the GeoMx IPA dataset for the area (C) and single-fiber ROIs (D) (P < 0.05; 95% confidence interval). (E and F) Volcano plots showing DEPs in unaffected (left) and LAMA2-CMD (right) area ROIs (E) and single-fiber ROIs (F), determined by 2-tailed mixed effects linear regression with FDR multiple test correction. The top horizontal line indicates the cutoff for proteins with an adjusted P value of less than 0.05. (G and H) LAMA2-CMD patient DEPs sorted into biological processes based on NanoString’s target group classification for area (G) and single-fiber ROIs (H), showing the 12 and 13 largest categories.
Age-dependent proteomic changes highlight progressive pathology in LAMA2-CMD muscle. For many patients with LAMA2-CMD, early symptoms can appear within the first week of life (20). Additionally, LAMA2-CMD symptoms, such as respiratory involvement and joint contractures, become more severe after the onset of adolescence (20). Therefore, we divided the dataset into younger and older patients, and their age-matched unaffected controls (Table 1). In the unaffected group, 4 DEPs were identified, while 18 enriched DEPs were found in the LAMA2-CMD younger patient ROIs (Figure 3A and Supplemental Tables 2 and 3). Notably, heat shock protein 70 (HSP70) and heat shock protein 27 (HSP27) were downregulated in the LAMA2-CMD group (Table 2). These molecular chaperones respond to stress stimuli, helping to maintain muscle fiber integrity and support muscle regeneration and recovery (21). The early loss of these proteins in patients with LAMA2-CMD may worsen skeletal muscle damage through protein accumulation and inhibit muscle regeneration over time.
 Figure 3
Figure 3Age-sorted protein expression data reveal protein accumulation in older patients with LAMA2-CMD. (A and C) Volcano plots showing differentially expressed proteins (DEPs) in area and single-fiber ROIs from the younger cohort, comparing unaffected and LAMA2-CMD tissues. (B and D) Volcano plots showing DEPs in area and single-fiber ROIs from the older cohort, comparing unaffected and LAMA2-CMD tissues, illustrating protein accumulation over time in the LAMA2-CMD skeletal muscle microenvironment and single fibers.
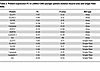 Table 2
Table 2Protein expression FC in LAMA2-CMD younger patient skeletal muscle area and single-fiber ROIs
In the older group, 322 DEPs were identified in the LAMA2-CMD area ROIs, while no enriched proteins were found in the unaffected group (Figure 3B). Collagen I showed the highest change, with a log2(fold change) (log2FC) of 3.0 (P = 0.021) in the older cohort ROIs but was not differentially expressed in the younger cohort, suggesting compensatory extracellular matrix (ECM) remodeling occurs over time (Figure 3, A and B). The single-fiber ROI datasets were also sorted by age, with younger and older patients compared to their age-matched controls (Figure 3, C and D). In younger patients, single myofibers displayed a mix of down- and upregulated DEPs (Figure 3C). Similar changes were also observed for the single-fiber ROIs compared with the area ROIs, primarily in the older group, where LAMA2-CMD skeletal muscle exhibited a disproportionate number of enriched DEPs — 113 in total — compared with zero DEPs in the unaffected group (Figure 3D). This large accumulation of proteins found in the muscle of older patients may be due to dysfunctional proteostasis due to high levels of cellular stress related to the severe muscle pathology, potentially due to the early loss of HSP70 and HSP27 (Figure 3a).
Pathway analysis reveals age-dependent oxidative stress, apoptosis, and immune dysregulation in LAMA2-CMD muscle tissue. Gene Ontology (GO) pathway analysis was performed on DEPs from the age-sorted data area ROIs with a log2FC cutoff of 0.25 (Figure 4, A–D). Analysis of downregulated proteins in the younger area ROIs revealed changes in LAMA2-CMD patient response to chemical and oxidative stress, apoptosis, and reactive oxygen species (ROS), among other biological processes (Figure 4A). In the single-fiber ROIs, both younger and older patients also showed an upregulation in ROS response and metabolic processes (Supplemental Figure 1). This may be linked to the early downregulation of key enzymes, superoxide dismutase 2 (SOD2) (log2FC = –1.04, P = 0.06) in the muscle fiber and skeletal muscle area, as well as catalase (log2FC = –1.25, P = 0.01) in the single-fiber ROIs, which are enzymatically responsible for breaking down superoxide and hydrogen peroxide, respectively (22) (Table 2). Analysis of upregulated proteins shows increased activation in inflammatory and adhesion mechanisms, as well as T cell activation. Notably, extrinsic apoptotic signaling pathway was enriched, suggesting LAMA2-CMD patient skeletal muscle fibers may prefer extrinsic apoptosis over intrinsic apoptosis (Figure 4B). GO analysis of the older muscle area ROIs had some similarities to the younger muscle area ROI expression profiles, such as downregulation of intrinsic apoptosis and upregulation of leukocyte modulation, T cell activation, and cell-to-cell adhesion regulation (Figure 4, C and D). Older patient downregulated biological processes were also involved in the regulation of skeletal muscle aging and activation of helper T cells (Figure 2C).
 Figure 4
Figure 4GO pathway analysis reveals altered biological processes as disease progresses in patients with LAMA2-CMD. (A and B) GO enrichment analyses of proteins with log2FC >0.25 for younger area ROI protein expression using down- and upregulated proteins, respectively. The 10 most down or upregulated biological processes are shown. (C and D) GO enrichment analyses of older area ROIs.
DSP analysis of dyW–/– mouse skeletal muscle highlights metabolic and immune dysregulation similar to human LAMA2-CMD. Using NanoString’s GeoMx Immuno-Oncology mouse panel, we performed DSP on wild-type (WT) and dyW–/– tibialis anterior (TA). The only commercially available mouse panel was much smaller than the human IPA panel. However, 50 of the 60 probes overlap with those in the human IPA panel (Figure 5A). ROIs were selected similarly to the human DSP, and the 60 different proteins were analyzed within the specified ROIs. Protein counts were normalized using negative controls (rabbit IgG, rat IgG2a, and rat IgG2b), and volcano plots were generated displaying DEPs of the area and single-fiber ROIs (Figure 5, B and C, and Supplemental Table 5). GO pathway analysis was performed on the downregulated and upregulated proteins between the dyW–/– and WT area ROIs (Figure 5, D and E). The downregulated biological processes indicate a dysregulation in lymphocyte development, as well as skeletal muscle metabolism. The downregulated proteins in the dyW–/– mouse involved in skeletal muscle metabolism include the biological processes of glucose, hexose, monosaccharide, and carbohydrate transmembrane transport (Figure 5D). Although the mouse protein panel is smaller than the human IPA panel, the upregulated biological processes matched 6 out of 10 biological processes in the younger patient area ROIs and shared 5 out of 10 upregulated biological processes found in the older patient area ROIs (Figure 5E). These upregulated biological processes include positive regulation of cell adhesion, leukocyte cell-to-cell adhesion, and T cell activation (Figure 4, B and D, and Figure 5E).
 Figure 5
Figure 5Digital spatial profiling of dyW–/– TA shows disrupted glucose metabolism and upregulated inflammatory processes. (A) Number of overlapping protein probes found in NanoString’s GeoMx human IPA panel and mouse immune-oncology panel. (B and C) Area and single-fiber ROI DEP volcano plots for WT compared to dyW–/–. (D and E) GO pathway analyses of proteins with log2FC >0.25 of down- and upregulated proteins. The 10 most down or upregulated biological processes are shown.
RhLAM-111 treatment restores laminin adhesion complex protein localization in dyW–/– mice. RhLAM-111 was produced and purified using proprietary techniques by Sarcomatrix and Catalent with codon-optimized laminin α, β, and γ cDNA chains. Quality control was performed by the Burkin lab. Shadow rotary imaging showed the rhLAM-111 had a cruciform structure, which has been previously demonstrated for the laminin-111 structure (23) (Figure 6A). Using gel electrophoresis and Coomassie staining techniques, we confirmed rhLAM-111 protein purity and individual chains were the correct sizes (Figure 6B).
 Figure 6
Figure 6Characterization of purified rhLAM-111. (A) Shadow rotary image of purified rhLAM-111. Scale bar: 200 nm. (B) Coomassie stain of rhLAM-111 run in 4%–20% SDS-PAGE gel, illustrating purification of therapeutic and verification of laminin chain sizes.
We have previously demonstrated that treatment with mouse or human laminin-111 prevents disease progression and improves viability in the LAMA2-CMD mouse model, the dyW–/– mouse (12, 24). Integrin-α7 (ITGA7) deficiency, a characteristic prevalent in patients with LAMA2-CMD and dyW–/– mice, led us to investigate the localization of protein components within laminin adhesion complexes. These include the dystrophin-glycoprotein complex (DGC) and α7β1-integrin. Using IF, we observed that, compared with WT tissue, laminin adhesion complex components such as ITGA7, α-dystroglycan (αDG), and α-sarcoglycan (SGCA) localization to the sarcolemma were reduced in dyW–/– mice (Figure 7A). In contrast, rhLAM-111 treatment restored these laminin adhesion complex proteins to the sarcolemma. This restoration was rapid and observed at 48 hours after treatment, with full restoration achieved within 7 days after treatment (Supplemental Figure 2). Using an antibody that specifically recognizes activated integrin-β1 (ITGB1), we found that dyW–/– muscle had little activated ITGB1 on the muscle fiber surface, which represents ECM engagement. RhLAM-111 treatment increased the presence of activated ITGB1 to nearly WT levels (Figure 7A). Interestingly, utrophin, which is typically localized at the myotendinous junction, was localized throughout the myofiber in dyW–/– muscle. After rhLAM-111 treatment, utrophin’s localization around myofibers was diminished, and the localization was restored to WT localization (Figure 7A). Together, these results suggest that rhLAM-111 can effectively restore the altered laminin adhesion complex localization and promote activation of ITGB1 in dyW–/– skeletal muscle.
 Figure 7
Figure 7RhLAM-111 restores laminin adhesion complex proteins’ localization and restores protein expression profile to WT levels. (A) Localization of protein components of laminin adhesion complexes in the dyW–/– muscle. Transverse sections of whole TA muscle from 5-week-old WT, dyW–/– and dyW–/– after 7 days rhLAM-111 treatment with antibodies against LAMA1, ITGA7, ITGB1, α-dystroglycan (αDG), α-sarcoglycan (SGCA), and utrophin (UTRN). Antibodies were visualized using indirect fluorescence microscopy. Scale bar: 50 μm. (B) Western blot analysis was performed to measure and quantify ITGA7, ITGB1, αDG, and SGCA (relative to total protein). For Western blotting, 40 μg of protein was loaded per well. n = 5 samples for WT and n = 4 for dyW–/– and dyW–/– + rhLAM-111. (C and D) Area and single-fiber ROI DEP volcano plots for dyW–/– compared to rhLAM-111–treated dyW–/–. Data are shown as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 by 1-way ANOVA followed by Bonferonni post-hoc comparison between all groups.
Western blot analysis in gastrocnemius muscle revealed substantial increases in laminin-adhesion complex proteins in dyW–/– mice compared with WT levels, including ITGA7 (P = 0.0013), ITGB1 (P = 0.0477), and αDG (P = 0.0037) (Figure 7B). In the contralateral rhLAM-111–treated leg, ITGA7 (P = 0.0006), ITGB1 (P = 0.0435), and αDG (P = 0.0011) protein levels were also significantly increased compared with WT (Figure 7B). Levels of SGCA were slightly increased in both the dyW–/– and dyW–/– + rhLAM-111 groups (Figure 7B). However, these differences were not statistically significant. Protein quantification was normalized to total protein, since common housekeeping controls such as GAPDH and actin can be altered in laminin-deficient muscle due to their roles in metabolism and cytoskeleton, respectively. Higher abundance of laminin-adhesion complex proteins may indicate a compensatory increase in protein production due to the inability of these proteins to anchor themselves within the sarcolemma properly. Previous studies have also indicated that DGC protein content is not a quality metric compared with the proper translocation to the sarcolemma (25, 26).
Restoration of signaling pathways and immune modulation in dyW–/– skeletal muscle following rhLAM-111 treatment. Laminin adhesion complexes play a crucial role in signal transduction of skeletal muscle. Although the DGC is a key mechanosensor in skeletal muscle, it also acts as a scaffold for signaling proteins (27). The transmembrane receptor α7β1-integrin is involved in cellular adhesion to the ECM and regulates numerous signaling pathways, including PI3K/AKT, MAPK, and TGF-β/SMAD pathways (28, 29). Given the disrupted localization of laminin adhesion complex components and downregulation of active ITGB1 in the dyW–/– mice, we hypothesized that these changes could lead to disruptions in major signaling pathways.
To explore these potential changes, we utilized NanoString’s GeoMx Immuno-Oncology protein panel to assess protein expression in the TA muscle of dyW–/– mice. Mice were intramuscularly treated at 4 weeks of age with 5 μg/kg bodyweight of rhLAM-111 in the left TA for 7 days, while the right leg served as a contralateral control and received an equal volume of phosphate-buffered saline (PBS). WT tissue was collected from littermates of the same age. Volcano plots were generated to compare the differences between dyW–/– and dyW–/– + rhLAM-111 area and single-fiber ROI DEPs (Figure 7, C and D).
In the area ROIs between the dyW–/– and dyW–/– + rhLAM-111 groups, there is a mixture of DEPs. Among the 12 DEPs enriched in the LAM group, 10 were also enriched in WT compared with dyW–/– (Figure 5B and Figure 7C). Two of the overlapping proteins, glucocorticoid-induced TNFR-related protein (GITR) and T cell immunoglobulin and mucin-domain containing-3 (TIM3), are involved in immune regulation. GITR enhances regulatory T cells, and TIM3 dampens T cell immune responses (30, 31). These results suggest that dyW–/– mice may experience modulation of the T cell immune response after rhLAM-111 treatment. Additionally, rhLAM-111 restores histone H3 protein levels in dyW–/– mice. In the human IPA panel, 13 different histone H3 targets display various posttranslational modifications, with 9 histone H3 markers having an FC decrease of over 20% (Supplemental Table 6). The downregulation of histone H3 in patients and dyW–/– mice may indicate that epigenetic dysregulation could be a hallmark of LAMA2-CMD pathology.
In the single-fiber ROIs, rhLAM-111 treatments appear to target both the MAPK and PI3K/AKT pathways. The most significant protein expression change observed was a prominent increase in phosphorylated c-Jun N-terminal kinase (p-JNK), with a log2FC of 3.66 (P = 0.00078) (Figure 7D). p-JNK acts as a molecular switch that, when activated, stimulates muscle fiber growth, primarily influencing fast-twitch fibers (32). Given JNK’s key role in the MAPK signaling network, we examined the expression of other MAPK proteins and observed low levels of phospho-mitogen-activated protein kinase kinase 1 (p-MEK1), phospho-extracellular signal–regulated kinases 1/2 (p-ERK1/2), and serine/threonine-protein kinase B-raf (p-BRAF) in dyW–/– in comparison with WT, in which rhLAM-111 treatment was able to increase and restore these proteins in the contralateral leg. RhLAM-111 treatment also increased proteins associated with the PI3K/AKT signaling network, including p-AKT1 at Ser473 (log2FC = 1.21, P = 0.066), phospho-glycogen synthase kinase 3A/B (p-GSKA/GSK3B) (log2FC = 2.01, P = 0.0090), and phospho-proline-rich AKT substrate 40 kDa (p-PRAS40) (log2FC = 2.01, P = 0.0031) (Figure 7D). p-AKT1 at Ser473 is required for the activation of the PI3K/AKT signaling pathway. p-AKT1 also promotes the activation of mTORC1 by facilitating the phosphorylation of downstream effectors such as PRAS40 (33). Another downstream target of AKT, p-GSK3A/GSK3B, is essential for glycogen metabolism and contributes to an anabolic state in skeletal muscle (34). However, there was also a significant increase in phospho-protein kinase, AMP-activated, α1 catalytic subunit (p-PRKAA1) (log2FC = 1.25, P = 0.0098), which initiates catabolic processes in skeletal muscle to generate ATP (35) (Figure 7D).
When comparing WT to rhLAM-111–treated dyW–/– single-fiber ROIs, WT expressed no enriched proteins, whereas rhLAM-111–treated samples showed 3 (Supplemental Figure 3). These findings suggest that rhLAM-111 treatment restores protein expression patterns similar to WT tissue after 7 days. The only enriched DEP between rhLAM-111 and WT was fibronectin (log2FC = 2.34, P = 0.033), which may be a result of the short-term treatment. Fibronectin levels may further be reduced with longer-term treatment.
RhLAM-111 treatment mitigates metabolic dysfunction and restores protein trafficking in early-stage LAMA2-CMD. DyW–/– mice were treated at 4 weeks of age and euthanized at 5 weeks, an age range chosen to better represent the younger cohort of patients in terms of disease progression and pathology. Consequently, we focused on protein expression levels and FCs in this younger cohort. The top 3 downregulated proteins identified in the younger ROI areas compared with the unaffected group were glucose transporter protein type 1 (GLUT1), ELAV-like RNA-binding protein 1 (ELAVL1), and insulin-like growth factor 1 receptor (IGF1R), all of which are critical to glucose metabolism and substrate availability in skeletal muscle (Table 2).
HSP27 and HSP70 showed reduced expression in the younger patients. IF was performed to look at the proteins’ expression patterns in the dyW–/– muscle and to explore laminin’s potential role. In dyW–/– TA muscle, HSP27 was localized to the membrane and was largely absent from the cytosol (Figure 8A). Following rhLAM-111 treatment, HSP27 exhibited cytosolic localization. HSP70 showed a similar pattern, with rhLAM-111 treatment inducing a substantial shift toward cytosolic localization (Figure 8A). The disrupted localization of HSP27 and HSP70 in laminin-α2–deficient muscle, combined with the disproportionately increased protein enrichment observed in older patients, highlights laminin’s critical role in regulating myofiber protein trafficking and expression.
 Figure 8
Figure 8RhLAM-111 treatment increases cytosolic heat shock protein expression and enhances GLUT1 localization to the sarcolemma after 7 days of treatment. (A) IF imaging of HSP27, HSP70, and GLUT1 in WT, dyW–/–, and dyW–/– + rhLAM-111 (contralateral legs). HSP27 and HSP70 are localized to skeletal muscle cytoplasm, which is associated with protein chaperone and unfolding activity. GLUT1 expression on the muscle fiber sarcolemma increases with rhLAM-111 treatments. Scale bar: 50 μm. (B) Western blot analysis was performed to measure and quantify HSP70 (relative to total protein). n = 4 per group. (C) Western blot quantification of GLUT1 (relative to total protein). n = 3 samples for WT and n = 4 for dyW–/– and dyW–/– + rhLAM-111 groups. (D) Enzyme histochemistry for NADH showing high levels of accumulation in PBS-treated dyW–/– TA compared with WT. RhLAM-111 restores NADH expression similar to WT levels in 7 days after treatment. Data are shown as mean ± SD. *P < 0.05; **P < 0.01 by 1-way ANOVA followed by Bonferonni post-hoc comparison between all groups.
GLUT1 localization was investigated using IF in WT and dyW–/– TA muscle. In dyW–/–, there was an absence of sarcolemmal GLUT1 localization (Figure 8A). However, in the rhLAM-111–treated contralateral leg, GLUT1 localization was restored to the sarcolemma. GLUT1 distribution also became more uniformly distributed around muscle fibers. These findings may suggest why there is a higher proportion of oxidative fiber types in LAMA2-CMD patient muscle, further elucidating a role for laminin in regulating metabolic function in muscle.
Protein levels of HSP70 and GLUT1 were significantly elevated in both dyW–/– and dyW–/– + rhLAM-111 skeletal muscle (Figure 8, B and C), a finding that contrasts with the human spatial proteomics data (Figure 2A and Table 2). HSP70 is a highly dynamic protein, and the molecular chaperone’s localization is critical for its biological activity (36). HSP70 is also highly expressed in macrophages and other inflammatory cells that are found in high abundance within the interstitial space of dyW–/– myofibers (37, 38). Western blot analysis was performed on whole muscle tissue lysate, whereas the patient’s spatial proteomic ROIs consist of single fibers and muscle fiber areas with low immune cell infiltrate and intact muscle fibers. Similarly, the elevated GLUT1 signal may reflect contributions from proliferative myofibroblasts, which rely heavily on aerobic glycolysis (39, 40). Inflammatory cells such as neutrophils and macrophages also express profuse levels of GLUT1 due to high metabolic demands (41–43). Altogether, these findings underscore that protein localization, rather than absolute abundance, provides the more biological relevance behind the pathomechanisms of dystrophic tissue.
GO pathway analysis of patient DEPs, as well as previous proteomic studies, demonstrated that LAMA2-CMD muscle has bioenergetic imbalances that contribute to constant oxidative stress and ROS accumulation (44). To assess metabolic activity, nicotinamide adenine dinucleotide (NADH) precipitate enzyme histochemistry was performed. Compared with WT, dyW–/– myofibers appeared much darker, and NADH accumulation was found throughout the entire fiber (Figure 8D). Within 7 days of rhLAM-111 treatment, NADH accumulation in the contralateral leg was reduced to levels comparable to WT. High levels of NADH do not directly indicate high levels of ROS, but rather the higher levels of NADH are indicative of impaired oxidative phosphorylation (OXPHOS) and therefore causes ROS accumulation. High NADH levels suggest a bottleneck in the mitochondrial electron transport chain, specifically at complex I, where NADH donates electrons (45).
Downregulated spatial omics proteins, HSP27, HSP70, and GLUT1, display altered localization in patients with LAMA2-CMD. HSP27 and HSP70 are highly dynamic molecular chaperone proteins that localize to different areas of skeletal muscle fibers in response to various stimuli (46). In unaffected patients, HSP27 can be found dispersed throughout the cytoplasm, where it binds to the cytoskeleton to help maintain structural integrity (Figure 9A). In response to mechanical stress or injury, HSP27 will rapidly translocate to the cell membrane, particularly near the Z-disks. In LAMA2-CMD, HSP27 can be seen heavily aggregated around the sarcolemma across varying ages and disease states (Figure 9A). Like HSP27, HSP70 is located intracellularly in the cytoplasm of myofibers; however, in response to injury and stress, HSP70 relocates to the sarcolemma (36, 46). By binding to the damaged proteins near the sarcolemma, HSP70 prevents them from aggregating and facilitates their repair. In unaffected tissue, HSP70 could be found heavily localized within the cytoplasm of myofibers (Figure 9B). However, similarly to HSP27, HSP70 was concentrated primarily around the myofibers of the 2-year-old (2-y.o.) and 17-y.o. patients (Figure 6B). In the 4-y.o. LAMA2-CMD patient, HSP70 IF staining appeared to be overall reduced and could be correlated with disease progression in patients with full laminin deficiency.
 Figure 9
Figure 9Patients with LAMA2-CMD exhibit altered localization of downregulated DSP proteins HSP27, HSP70, and GLUT1. (A–C) IF imaging of HSP27, HSP70, and GLUT1 in unaffected and LAMA2-CMD muscle biopsies. *The 17-y.o. patient has a partial laminin deficiency. Scale bar: 50 μm.
GLUT1 localization was also investigated using IF in the patient tissues. In the unaffected controls, GLUT1 could be found uniformly distributed around the myofibers (Figure 9C). However, GLUT1 staining was almost completely diminished in the 2-y.o. and 4-y.o. LAMA2-CMD patients. Partial GLUT1 staining could be seen in the 17-y.o. patient tissue. This partial staining could potentially be correlated with the patient’s partial laminin deficiency, and GLUT1 could be localized to the sarcolemma in regions where the laminin is present (Figure 9C).
Cytosolic p-JNK localization decreases alongside disease progression in LAMA2-CMD muscle. Since p-JNK exhibited the largest FC with high significance, IF was performed to observe the localization in mice (Figure 10A). Higher levels of p-JNK were localized in the cytosol of WT and dyW–/– + rhLAM-111 mouse myofibers compared with dyW–/–. In human studies, no significant changes in JNK1/JNK2/JNK3 expression was detected in younger patients. However, the IPA panel used in these studies measured only total JNK, not the activated phosphorylated form of JNK used in the mouse panel.
 Figure 10
Figure 10Cytosolic localization of p-JNK in dyW–/– and LAMA2-CMD skeletal muscle. (A) IF of p-JNK in WT, dyW–/–, and dyW–/– + rhLAM-111. p-JNK localization in the cytoplasm is decreased in the dyW–/– muscle and rhLAM-111 treatments increase cytosolic p-JNK after 7 days. Images on the bottom row are with wheat germ agglutinin (magenta) to highlight the muscle fiber sarcolemma. Scale bar: 50 μm. (B) IF of p-JNK in patients with LAMA2-CMD and age-matched controls show a decreasing presence of p-JNK in the cytosol as patients age. Scale bar: 50 μm.
Next, IF was utilized to observe the localization of p-JNK in patients with LAMA2-CMD. LAMA2-CMD patients ages 2, 3, and 7 y.o. were included in the panels, with unaffected controls 3, 3, and 8 y.o. (Figure 10B). In the 2-y.o. patient, p-JNK was present in the cytosol in some muscle fibers; however, in the 3-y.o. patient, p-JNK began to migrate to the sarcolemma of the myofibers, with some p-JNK present in the peripheral cytoplasm. The 7-y.o. patient’s cytosolic p-JNK was almost completely diminished, and some fibers had p-JNK strongly localized to the sarcolemma. Meanwhile, in the unaffected control tissue, p-JNK was present in the cytosol in most fibers, suggesting cytosolic p-JNK may be present in a preferred muscle fiber type.












-
Discussion
In this study, we used spatial proteomics combined with comprehensive pathway analyses to demonstrate that profound changes in laminin-binding adhesion-signaling complexes play a crucial role in disease progression in patients with LAMA2-CMD and the dyW–/– mouse model. These findings contribute to our understanding of how the loss of crosstalk between laminin and its adhesion complexes contributes to disease pathology and highlight potential targets for therapeutic intervention.
Using data from spatial proteomics and IF, we propose a mechanism of action by which laminin alleviates ROS accumulation through α7β1-integrin (Figure 11). Patients with LAMA2-CMD can experience secondary α7β1-integrin, and in the absence of laminin, α7β1-integrin remains in an inactive closed state and is not properly localized to the sarcolemma, preventing downstream signaling (47–49). When rhLAM-111 enters the basal lamina, it binds to integrin-α7, which then activates integrin-β1 (Figure 7A). Integrin-β1 regulates several proteins, including caveolin-1, GLUT1, and IGF1R, all of which are found to be downregulated in patients with LAMA2-CMD (Table 2) (50–52). Upon activation, integrin-β1 activates the proto-oncogene tyrosine-protein kinase (Src), which directly phosphorylates caveolin-1; this activation influences caveolin-1’s stabilization and downstream signaling (53). Activating caveolin-1 facilitates the translocation of GLUT1 and IGF1R to the cell membrane, enabling glucose influx and adaptation to hypoxic conditions. Restoration of the laminin adhesion complexes also promotes proper mechanotransduction signaling via JNK, which is directly phosphorylated and activated by Src. This in turn enhances c-Jun transcription, promoting glycolytic activity and enhancing GLUT1 expression (54–57). GLUT1 plays a key role in regulating ROS, unlike GLUT4, which has no such association (58). Activated JNK undergoes continuous autophosphorylation in response to ROS recognition, while a metabolic switch from oxidative to glycolytic further reduces ROS accumulation (59, 60). This shift alleviates muscle stress signals, leading to the dispersion of protein accumulation or stress granules. RNA stabilization, mediated by the RNA-binding protein ELAVL1, is supported by the activity of chaperone proteins HSP27 and HSP70, which aid in the unraveling process within the cytosol (61, 62). The increase in cytosolic HSP27 and HSP70 inhibits the JNK-mediated route of apoptosis through binding to the adaptor molecule Daxx (63, 64).
 Figure 11
Figure 11Schematic of molecular interactions between dysregulated spatial proteomics proteins’ biological processes and related pathways. All proteins included in the schematic are disrupted in human or mouse spatial proteomics and IF. Molecular interactions and associations are assumed based on peer-reviewed literature.
Analysis of younger patients with LAMA2-CMD identified early downregulation of molecular chaperones HSP27 and HSP70, proteins critical for stress response and muscle regeneration (21) (Table 2). The loss of these chaperones likely exacerbates skeletal muscle damage and impairs regenerative properties, further advancing disease progression. The reduction in antioxidant enzymes, such as SOD2 and catalase, may heighten oxidative stress in younger patients, contributing to early muscle fiber damage and apoptosis. Older patients with LAMA2-CMD displayed elevated accumulation of many DEPs compared with healthy controls, suggesting problems with muscle fiber autophagy, a known issue for DMD (65) or regulation of proteostasis. Downregulation of proteins is critical for maintaining ROS, plus protein accumulation over time may indicate stress granule formation within the myocytes due to oxidative stress found in patients with LAMA2-CMD and dyW–/– mice (66, 67). Our studies revealed that activated JNK in LAMA2-CMD patient tissue became more deficient in the cytosol as patients age. Typically, elevated levels of ROS are associated with JNK activation, but in patients with LAMA2-CMD and dyW–/– mice, this feedback mechanism appears to be dysregulated (68). Since cytosolic p-JNK is restored through rhLAM-111 treatments, JNK signaling may be regulated or initiated through laminin adhesion complexes.
Previous proteomic and transcriptomic studies have similarly observed deficiencies in LAMA2-CMD muscle metabolism, resulting from the downregulation of numerous proteins involved in OXPHOS and glycolysis (44, 69–71). Furthermore, the use of antioxidants in the dy2J/dy2J LAMA2-CMD mouse model decreases inflammation and oxidative stress in skeletal muscle (72). Our spatial proteomics findings align with these previous studies, revealing deficiencies in glycolysis-related proteins, as well as key mitochondrial antioxidant enzymes SOD2 and catalase in the younger patient tissue. The 3 greatest FC reductions were in glycolysis-related proteins: GLUT1, ELAVL1, and IGF1R. GLUT1 facilitates glucose uptake in the cell and is a rate-limiting step in the process of glycolysis (43, 73). IGF1R also plays a marked role in glycolysis regulation and is further involved in muscle regeneration (74). Studies have shown that overexpression of a precursor for muscle-specific IGF-1 improved muscle regeneration and longevity in laminin-α2–deficient mice, highlighting the importance of restoring the glycolytic ability of laminin-deficient muscle (75). On the other hand, ELAVL1 is not directly involved in glycolysis, but plays a role in skeletal muscle metabolic flexibility and stabilizes mRNA of many important glycolytic proteins, including GLUT1 (76, 77). Reduced expression of these proteins likely leads to reduced myofiber glucose availability and slow-twitch fiber-type prevalence observed in patients with LAMA2-CMD (78). Altogether, these findings underscore how metabolic deficiencies in laminin-deficient muscle contribute to disease pathology and highlight metabolic restoration as a promising therapeutic.
In the dyW–/– mouse model, rhLAM-111 treatment demonstrated the ability to restore key components of the laminin adhesion complex, including ITGA7, active ITGB1, and αDG, to their normal cellular localization. Restoration of the localization of proteins is likely critical for proper signal transduction and structural integrity of the muscle. Western blot analysis revealed significant increases in the protein levels of ITGA7, ITGB1, and αDG in dyW–/– compared with WT mice. These proteins may not be properly localized to the sarcolemma without the presence of laminin, and therefore, myofibers may produce more proteins in order to compensate for the laminin adhesion complex proteins. Protein quantification of HSP70 and GLUT1 was also elevated in the dyW–/– mice, which could be associated with fibroblasts and immune cells found in the dystrophic tissue. Previous studies have demonstrated that the localization of the DGC is a better outcome measure than protein quantification (25, 26). Therefore, spatial omics studies using defined ROIs, in combination with IF analysis, can provide valuable insights into the molecular and cellular events occurring within dystrophic skeletal muscle.
RhLAM-111 treatment effectively restored MAPK and PI3K/AKT signaling pathways, which are both essential for muscle growth, apoptotic regulation, and metabolic balance. Increased p-JNK and p-AKT1 suggest that rhLAM-111 treatment promotes an anabolic state beneficial to muscle repair while maintaining the myofiber energy balance via the coactivation of catabolic p-PRKAA1 (32, 79, 80). The restoration of histone H3 protein levels also suggests that rhLAM-111 may play a role in epigenetic dysregulation, a potential hallmark of LAMA2-CMD pathology.
While our findings elucidated signaling pathway and metabolic dysregulation in laminin-α2–deficient muscle alongside the therapeutic effects of rhLAM-111, several limitations are acknowledged. First, our study included a relatively small cohort of patients with LAMA2-CMD and age-matched unaffected controls for DSP and should be expanded to validate our findings. The limited availability of patient tissue constrained the power of our analyses, leading us to instead focus on effect sizes and biological relevance. Second, the dyW–/– mice were treated with rhLAM-111 for only 7 days, which may represent only the initial DEP changes and limit our understanding of the benefits or limitations of long-term treatment. For example, Western blot analysis showed significantly higher amounts of protein in both dyW–/– groups compared with WT levels. However, 7 days may not be a sufficiently long treatment time to be able to observe any possible stabilization of protein levels with rhLAM-111 administration. Additionally, this short-term treatment window may not reveal any potential immune response that the dyW–/– mouse could develop from an adaptive immune response to a recombinant human protein. There is also always a possibility for an immune response to develop to a large dose of protein therapeutic in patients due to potential glycosylation changes between native and recombinant protein isoforms and innate immune activation. Third, the NanoString GeoMx panels used in this study contained a limited number of probes, particularly in the mouse panel, which limits our ability to perform comparisons to patients with LAMA2-CMD. Expanding the DSP panels to include additional protein targets could provide a more robust investigation of the similarities and differences between the LAMA2-CMD human and mouse model signaling pathways.
In conclusion, this study advances our understanding of LAMA2-CMD by elucidating the complex interplay of dysregulated proteostasis, oxidative stress, and metabolic deficiencies in both human patients and the dyW–/– mouse model. Using spatial proteomics and pathway analyses, we identified key downregulated DEPs in younger patients with LAMA2-CMD, including HSP27, HSP70, and caveolin-1, which all play a key role in protein trafficking, folding, and stabilization. Additionally, antioxidant enzymes SOD2 and catalase were also downregulated. Further studies in dyW–/– mice illustrated the dysregulation of the laminin adhesion complexes, which were later restored with rhLAM-111 treatments, including the activation of ITGB1. DSP mouse studies identified several rescued signaling proteins, including p-JNK, p-GSK3A/GSK3B, and p-PRKAA1, all of which play crucial roles in skeletal muscle metabolism. The improper localization of GLUT1 and pathway analysis in the human studies also indicated increased oxidative stress and decreased glycolysis. Our spatial proteomics findings align with previous transcriptomic and proteomic human and mouse studies, which have also described mitochondrial bioenergetic impairment and ROS vulnerability (44, 69, 71, 72, 81). Increased NADH accumulation has been observed in LAMA2-CMD patient tissue (63), prompting us to assess NADH levels in the dyW–/– mice using immunohistochemical tissue staining. DyW–/– muscle also exhibited abnormal accumulation of NADH, which could be attenuated after 7 days of rhLAM-111 treatment in the contralateral leg.
These findings underscore the contribution of laminin-associated pathways to muscle disease progression and highlight new potential therapeutic targets for LAMA2-CMD. RhLAM-111’s therapeutic potential is highlighted through its ability to restore key adhesion-signaling complexes, such as the activation of α7β1-integrin, its influence over the localization of protective molecular chaperones, and its promotion of a more glycolytic state. These findings pave the way for future studies exploring the clinical application of rhLAM-111 and other laminin-based therapies to mitigate disease progression and improve outcomes in LAMA2-CMD.



