Research ArticleGastroenterologyImmunologyInflammation
Open Access | ![]() 10.1172/jci.insight.178595
10.1172/jci.insight.178595
Unique epithelial proliferative transcriptomic signature in proton pump inhibitor–responsive pediatric eosinophilic esophagitis
Somdutta Chakraborty,1 Ankit Sharma,1 Sahiti Marella,1,2 Christian F. Rizza,1,2 Patrick A. O’Brien,1,2 Varsha Ganesan,1 Gila Idelman,1 Susie Min,3 Mayee Chen,1 Talaya McCright-Gill,4 Nancy Gonzalez,4 Alexandros D. Polydorides,5 Paul S. Foster,6 Simon P. Hogan,1,2 and Mirna Chehade4
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Chakraborty, S. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by
Sharma, A.
in:
PubMed
|
Google Scholar
|

1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Marella, S. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Rizza, C. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by O’Brien, P. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Ganesan, V. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Idelman, G. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Min, S. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Chen, M. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by McCright-Gill, T. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Gonzalez, N. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Polydorides, A. in: PubMed | Google Scholar
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by
Foster, P.
in:
PubMed
|
Google Scholar
|
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by
Hogan, S.
in:
PubMed
|
Google Scholar
|
1Mary H. Weiser Food Allergy Center,
2Molecular Pathology Graduate Program, Department of Pathology, and
3Department of Internal Medicine, Michigan Medicine, University of Michigan, Ann Arbor, Michigan, USA.
4Mount Sinai Center for Eosinophilic Disorders and
5Department of Pathology, Molecular and Cell-Based Medicine, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
6Faculty of Medicine, Health and Human Sciences, Woolcock Institute of Medical Research, Macquarie University, New South Wales, Australia.
Address correspondence to: Mirna Chehade, Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1198, New York, New York, 10029, USA. Phone: 212.241.4880; Email: mirna.chehade@mssm.edu. Or to: Simon P. Hogan, Mary H Weiser Food Allergy Center, Department of Pathology, Michigan Medicine, University of Michigan, 109 Zina Pitcher Place, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.9923; Email: sihogan@med.umich.edu.
Authorship note: SC, AS, and SM contributed equally to this work.
Find articles by Chehade, M. in: PubMed | Google Scholar
Published October 8, 2025 - More info
JCI Insight. 2025;10(19):e178595. https://doi.org/10.1172/jci.insight.178595.
© 2025 Chakraborty et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: December 27, 2023; Accepted: July 25, 2025
-
Results
Characteristics of PPI-R and PPI-UR patients with EoE. Ten pediatric patients with EoE (age, 10.4 ± 3.4 years; M/F, 6:4) underwent esophagogastroduodenoscopy (EGD) with biopsies and were diagnosed with EoE per consensus guidelines (23). Patients underwent PPI monotherapy (omeprazole or lansoprazole) for 8–12 weeks followed by repeat EGD with biopsies. PPI adherence for all patients was confirmed by the treating clinician prior to their repeat endoscopies. Of the n = 10 patients, 5 individuals demonstrated PPI-R, and 5 individuals were unresponsive to PPI monotherapy (Table 1, Table 2, and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.178595DS1). We stratified the individuals into either PPI- R or PPI-UR groups and recruited participants matched for age, sex, and atopic status (age, 7.2 ± 3.5 years; M/F, 3:2) assigned as controls, who underwent EGD with biopsies. Participants acting as controls had 0 esophageal eos/HPF on biopsies and absent endoscopic findings inclusive of any EoE features on endoscopy, no esophageal or gastrointestinal pathology, and no other gastrointestinal diseases (Table 2 and Supplemental Table 1). Demographics and clinical, endoscopic, and histologic characteristics of the PPI-R and PPI-UR patients at diagnosis and following PPI monotherapy and the individuals acting as controls are described in Tables 1 and 2 and Supplemental Table 1. At diagnosis, the peak for esophageal eos/HPF (across all biopsy sites) was 94.2 ± 41.3 for the PPI-R group and 134.4 ± 49.6 for the PPI-UR group. Following PPI therapy, peak esophageal eos/HPF (across all biopsy sites) in the PPI-R group was 3.6 ± 2.6 and 125.6 ± 61.0 in the PPI-UR group. In the distal esophagus, where biopsies for RNA-Seq analyses were obtained, peak esophageal eos/HPF was 87.8 ± 46.9 for the PPI-R group and 132.2 ± 48.7 for the PPI-UR group at diagnosis. Following PPI therapy, peak distal esophageal eos/HPF in the PPI-R group was 3.0 ± 1.9 and 66.6 ± 46.0 in the PPI-UR group (Table 2). We observed statistically significant improvement in endoscopic (endoscopic reference score [EREFS]) and histologic (EoE histologic scoring system [EoE-HSS]) outcomes in PPI-R patients with EoE following PPI therapy (EREFS, total distal score, 3.4 ± 0.9 vs. 0.8 ± 0.8; EoE-HSS, distal grade score, 0.55 ± 0.14 vs. 0.12 ± 0.11; data represented as diagnosis vs. after PPI therapy, P < 0.01) (Table 2 and Supplemental Table 1). The PPI-R and PPI-UR groups had no statistically significant differences in mean age, M/F ratio, atopic status, and peak esophageal eos/HPF, EREFS, and EoE-HSS at diagnosis (Tables 1 and 2).
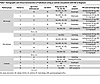 Table 1
Table 1Demographic and clinical characteristics of individuals acting as controls and patients with EoE at diagnosis
 Table 2
Table 2Histological and endoscopic parameters in individuals acting as controls and patients with EoE at diagnosis and following PPI therapy
RNA-Seq analyses at diagnosis. A total of 25 esophageal biopsy samples from n = 5 PPI-R patients with EoE (at diagnosis [before PPI] and following 8- to 12-week PPI therapy), n = 5 PPI-UR patients with EoE (at diagnosis [before PPI] and following 8- to 12-week PPI therapy), and 5 healthy individuals acting as controls were collected and analyzed by RNA-Seq (Figure 1A). Data processing, normalization, and quality control resulted in n = 43,186 transcripts with at least 1 raw read in any 1 sample of any group that mapped to >98% human reference genome Grch38. We observed an average of 34,122 raw read counts in the control samples, with 27,634 (82.48%) common genes expressed in each sample; an average of 21,446 raw read counts with 17,768 (82.8%) common genes expressed among PPI-R individuals; and an average of 23,521 raw read counts, with 18,343 (72.8%) common genes expressed among PPI-UR individuals.
 Figure 1
Figure 1RNA-Seq analysis of PPI-R and PPI-UR patients with EoE at diagnosis. (A) Summary schematic of 25 esophageal biopsy samples (n = 5 PPI-R at diagnosis, n = 5 PPI-R following PPI therapy, n = 5 PPI-UR at diagnosis, n = 5 PPI-UR following PPI therapy, and n = 5 healthy controls). (B and C) Comparative gene expression of DEGs between treatment-naive (B) individuals acting as healthy controls and PPI-R patients with EoE (FDR < 0.05; log2FC > 1) and (C) individuals acting as healthy controls and PPI-UR patients with EoE (FDR < 0.05; log2FC > 1). (D and E) Comparison of enriched biological processes and pathways in (D) individuals acting as healthy controls and PPI-R patients with EoE and (E) individuals acting as healthy controls and PPI-UR patients with EoE. (F) Heatmap of enriched genes associated with the immune and inflammatory response in individuals acting as healthy controls and PPI-R and PPI-UR patients with EoE at diagnosis. (G and H) Heatmap of enriched pathways (upregulated and downregulated) in individuals acting as healthy controls and PPI-R and PPI-UR patients with EoE at diagnosis.
We performed comparative gene expression analysis between individuals acting as controls and PPI-R and PPI-UR individuals with EoE at diagnosis using pairwise comparison cutoff criteria of FDR < 0.05; log2fold change (log2FC) > 1. We identified a total of 2,296 (1,022 upregulated and 1,274 downregulated) differentially expressed genes (DEGs) between healthy individuals acting as controls and PPI-R individuals with EoE and 4,084 (1,929 upregulated and 2155 downregulated) between individuals acting as controls and PPI-UR individuals with EoE (Figure 1, B and C, and Supplemental Table 2, A and B). DAVID pathway analyses revealed common dysregulation of inflammatory response and immune response pathways, in particular dysregulation of pro–type 2 genes previously identified as part of the EoE transcriptome (CCL26, CCL24, TNAIP6, ALOX15, FFAR3, IL13) as well as additional genes associated with chemotaxis (CCL13, CCL18, CCL23, CXCL1, CXCL6, and CXCL8), CD4+ type 2 immune genes (TNFRSF4, PTGER2), and IL-20 cytokine family members (IL19 and IL26) (Figure 1, D–F; Supplemental Table 3, A and B; and Supplemental Table 4A). Notably, the immune and inflammatory response genes were markedly more dysregulated (higher expression of corresponding genes) in PPI-UR individuals with EoE than in PPI-R individuals with EoE at diagnosis (Figure 1F and Supplemental Table 4A). Examination of the DEGs and most significantly enriched pathways within PPI-R EoE and PPI-UR EoE groups at diagnosis revealed significant upregulation of pathways involved in “cellular response to LPS,” “defense response to virus,” “IFN-gamma-mediated signaling,” and “type I IFN signaling pathways,” and downregulation of “cellular response to zinc ion,” “triglyceride homeostasis,” and “xenobiotic metabolic process” pathways in PPI-R EoE (Figure 1, D and G; Supplemental Table 3A; and Supplemental Table 4B). In contrast, the most enriched dysregulated pathways observed in the PPI-UR EoE group at diagnosis were associated with “apoptotic signaling pathways,” “positive regulation of chemokine production GTPase activity,” “IL6 production,” and “sister chromatid cohesion” (Figure 1, E and H; Supplemental Table 3B; and Supplemental Table 4C). The genes associated with the enriched pathways (upregulated and downregulated) were significantly more dysregulated in the PPI-UR group than the PPI-R group (Figure 1, F–H). To validate our RNA-Seq analyses, we compared our RNA-Seq data with previously reported transcriptomic profiling analyses performed on adult and pediatric EoE esophageal biopsies (GSE58640, GSE197702) (24). We showed that there was significant overlap in our PPI-R and PPI-UR EoE groups at diagnosis with that described in adult and pediatric EoE esophageal biopsies (Supplemental Figure 1A). Furthermore, when comparing the FC of DEGs between studies, we found a significant correlation in expression of DEGs within the external datasets and PPI-R and PPI-UR EoE at diagnosis (Supplemental Figure 1B). Collectively, these studies suggest common and unique dysregulated genes and pathways between PPI-R EoE and PPI-UR individuals with EoE at diagnosis and that there are conserved molecular signatures in EoE regardless of age group or PPI-R.
We next examined the involvement of the hematopoietic cell compartment in PPI-R and PPI-UR EoE at diagnosis. To do this, we mapped hematopoietic cell–specific gene markers identified using the PangloDB database (http://panglaodb.se) onto the normalized gene expression profile of PPI-R and PPI-UR EoE transcriptome at diagnosis. We identified high expression of mast cell (CMA1, CPA3, CKIT, CTSG, and RGS13), eos (EPX and IL5RA), and CD4+ Th2 cell (AHR, GATA3, IL13, IL17RB) signature genes both in PPI-R EoE and PPI-UR EoE at diagnosis (Figure 2A and Supplemental Table 5, A–C). Overall, the type 2 immune cell gene signature was enhanced in the treatment-naive PPI-UR EoE group compared with PPI-R EoE group (Figure 2A). Notably, we observed enrichment of a neutrophil (ITGAM, LCN2, NCF1, S100A8, and TLR2) and B cell signature (IGHD, IGLC3, CD52, IGLC2, CD79A, and CD19) in the PPI-UR EoE group that was not observed in PPI-R EoE at diagnosis (Figure 2A and Supplemental Table 5, A–C). To validate these observations, we performed immunofluorescence and histochemistry analyses on esophageal biopsies from PPI-R and PPI-UR individuals with EoE at diagnosis staining for mast cells (anti-tryptase) and evidence of esophageal epithelial proliferation (anti-Ki67). We observed that Tryptase+ mast cell numbers were significantly increased in both PPI-R and PPI-UR EoE compared with controls at diagnosis (Figure 2, B and C).
 Figure 2
Figure 2Hematopoietic and esophageal epithelial transcriptomes in PPI-R and PPI-UR EoE at diagnosis. (A) Heatmap of the gene expression profile of hematopoietic cell–specific markers in individuals acting as healthy controls and PPI-R and PPI-UR patients with EoE at diagnosis. (B) IHC-stained esophageal biopsies from PPI-R and PPI-UR patients showing tryptase+ mast cells (inset). (C) Quantifications of mast cells per unit area of biopsied tissue in individuals acting as healthy controls and PPI-R and PPI-UR patients (n = 5 per group). (D) Heatmap of the gene expression profile of esophageal epithelial cell type–specific markers in individuals acting as healthy controls and PPI-R and PPI-UR patients with EoE at diagnosis. (E) Confocal images of esophageal biopsies of individuals acting as healthy controls and PPI-R and PPI-UR patients, showing nuclei (DAPI) and proliferating cells (Ki67). Basement membrane is traced in white. (F) Quantifications of proliferating cells along the basement membrane in individuals acting as healthy controls and PPI-R and PPI-UR patients (n = 5 per group). (C and F) 1-way ANOVA was used to determine significance between the groups; *P < 0.05, **P < 0.01, ****P < 0.0001.
Utilizing the gene signatures derived from single-cell RNA-Seq (scRNA-Seq) analyses of esophageal epithelial populations (GSE201153) (25), we examined esophageal epithelial cell population gene expression in PPI-R and PPI-UR EoE at diagnosis. We identified the presence of quiescent and proliferating basal cells, Trans1 and Trans2 suprabasal cells, and low and high differentiated squamous epithelial cell transcriptomes in control individuals (Figure 2D and Supplemental Table 6). Consistent with a predominant mature squamous esophageal epithelium, differentiated squamous epithelial cell genes were highly enriched as compared with suprabasal and basal cell genes in esophageal biopsy samples from control individuals (Figure 2D and Supplemental Table 6). Notably, we observed dedifferentiation of the esophageal epithelial layer in both PPI-R and PPI-UR EoE at diagnosis as indicated by decreased expression of low differentiated genes (WDR26, WNK1, PADI1, and TGM3) and high differentiated (MT1G, MT1E, MT1H, HLPDA, and KRTAP3) esophageal epithelial cell genes. Furthermore, we observed enriched expression of proliferating epithelial genes (HMGB2, H2AFZ, TUBA1B, STMN1, UBE2C, TOP2A, RRM2, and CCNB1) in PPI-UR EoE at diagnosis, suggesting increased esophageal epithelial proliferation. Intriguingly, PPI-UR EoE at diagnosis had greater expression of proliferating epithelial genes (HMGB2, H2AFZ, TUBA1B, STMN1, UBE2C, TOP2A, RRM2, and CCNB1) as compared with PPI-R EoE (Figure 2D and Supplemental Table 6). Ki-67 staining of distal esophageal biopsies of pediatric individuals revealed that the level of epithelial proliferation was significantly higher in PPI-R and PPI-UR patients with EoE compared with that in individuals acting as controls at diagnosis (Figure 2, E and F). While the number of Ki-67+ esophageal epithelial cells was higher in the PPI-UR EoE group compared with PPI-R EoE group, the level of epithelial proliferation was not significantly different between groups. Collectively, these data support common CD4+ Th2 cell, eos and mast cell inflammatory signature, and dedifferentiation of the esophageal epithelium in PPI-R and PPI-UR EoE at diagnosis and that PPI-UR EoE at diagnosis was associated with enrichment of esophageal proliferative gene signature.
Relationship between gene expression and endoscopic severity. We next examined if any DEGs correlated with endoscopic parameters (EREFS score) in PPI-R and PPI-UR EoE at diagnosis. We show that the EREFS score of the distal esophagus was significantly elevated in PPI-R and PPI-UR EoE at diagnosis and that PPI therapy significantly reduced the EREFS score in PPI-R EoE but not in PPI-UR EoE (Table 2 and Supplemental Figure 2A). Correlation analyses revealed a positive correlation between distal esophageal histologic parameters (peak eos/HPF, BZH, DIS, and grade total score) and EREFS total distal score (peak count P = 0.002; r = 0.72, DIS P = 9.51 × 10–6; r = 0.89, BZH P = 6.87 × 10–7; r = 0.92 and total grade score P = 1.93 × 10–5; r = 0.88) (Supplemental Figure 2B and Supplemental Table 7). Correlation analysis between endoscopic parameter (total distal score) and DEGs in PPI-R and PPI-UR EoE at diagnosis identified that 2,064 of 2,296 genes correlated with the total distal EREFS score in PPI-R EoE and 3,527 of 4,084 genes correlated with total distal EREFS score in PPI-UR EoE (Supplemental Figure 2C and Supplemental Table 7). Gene ontology (GO) analysis analyses identified enrichment of genes associated with biological processes, including “inflammatory response” and “detoxification of copper ion”, which correlated with EREFS score in both PPI-R and PPI-UR EoE groups. However, biological processes such as “type I IFN signaling pathway,” “defense response to virus,” and “apoptotic signaling pathway,” uniquely correlated with EREFS score in PPI-R EoE and “cornification,” “chromosome segregation,” and “mitotic spindle organization” were specific for PPI-UR EoE (Supplemental Figure 2D).
Relationship between gene expression and histological characteristics. We next examined the relationship between DEGs in PPI-R EoE and PPI-UR EoE at diagnosis and histologic alterations, including eos/HPF, BZH, and DIS. To do this we performed a flatten matrix correlation analyses of 2,296 and 4,084 DEGs in PPI R- and PPI-UR EoE at diagnosis with the histological parameters of the distal esophagus determined using the EoE-HSS score criteria (26). We identified that 681 of 2,296 DEGs in PPI-R individuals with EoE correlated with peak eos/HPF count; 1,421 of 2,296 DEGs correlated with DIS; 1,745 of 2,296 DEGs correlated with BZH; and 1,332 of 2,296 DEGs correlated with the distal EoE-HSS total grade score. To get insight into the biological processes that contribute to the histologic alterations, eos/HPF, BZH, DIS, and distal EoE-HSS grade total score, we performed GO analyses on the DEGs that correlated with the individual histologic characteristics (Figure 3). We show that the biological processes “defense response to virus” and “apoptotic process” were enriched with peak eos/HPF count, BZH, and total grade score. Furthermore, the biological processes “inflammatory response” and “type I IFN signaling pathway” were enriched in BZH and total grade score, suggesting that these biological processes contribute to these histologic characteristics (Figure 3 and Supplemental Tables 8 and 9). Intriguingly, DEGs that correlated with DIS in PPI-R EoE were enriched for the biological processes associated with “detoxification of copper ion,” “defense response to virus,” “negative regulation of growth,” “cellular zinc ion homeostasis,” and “cellular response to copper ion,” suggesting that processes that regulate DIS are distinct from those associated with eos/HPF and BZH (Figure 3A and Supplemental Table 9, A–D). The DEGs that dominated the signature were the metallothionein (MT) genes (MT2A, MT1L, MT1M, MT1F, MT1G), which are low-molecular-weight metal binding proteins that play an important role in regulating metal homeostasis and controlling physiological heavy metal toxicity, DNA damage, and oxidative stress (27). In PPI-UR EoE, of the 4,084 DEGs, 1,544 DEGs correlated with distal peak eos count; 2,201 DEGs correlated with DIS; 3,113 correlated with BZH; and 2,618 correlated with distal grade total score (Supplemental Table 9, E–H). In UR EoE, there was overlap in the enriched biological processes, such as “mitotic spindle organization” and “chromosome segregation,” that correlated with peak eos/HPF count, BZH, and total grade score (Figure 3B). Furthermore, there were common biological processes, including “mitotic spindle organization,” “chromosome segregation,” and “antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent,” that correlated between BZH and total grade Score. Notably, the top enriched biological processes that correlated with the histologic characteristics, peak eos/HPF count, BZH, and total grade score in PPI-R EoE and PPI-UR EoE at diagnosis were different. The biologic processes “defense response to virus,” “inflammatory response,” and “type I IFN signaling pathway” were dominant in PPI-R EoE whereas “mitotic spindle organization” and “chromosome segregation” was most enriched in PPI-UR EoE (Figure 3 and Supplemental Table 9, A–H). The top biologic processes that correlated with DIS in UR EoE had the least overlap with the other histologic characteristics, peak eos/ HPF count, BZH and total grade score. Interestingly, the most enriched biologic processes in PPI-UR EoE were like those observed in PPI-R EoE, including “negative regulation of growth” and “cellular zinc ion homeostasis,” that possessed a strong MT gene signature and supported a role for these processes in DIS in both PPI-R and PPI-UR EoE (Figure 3 and Supplemental Table 9, A–H).
 Figure 3
Figure 3Relationship between gene expression and endoscopic severity in PPI-R and PPI-UR EoE at diagnosis. (A) DEGs and associated pathways that correlated with peak eosinophil count, DIS, BZH, and total histological score in PPI-R patients with EoE at diagnosis. Histograms represent a flatten matrix correlation analyses of DEGs in PPI R- and PPI-UR EoE at diagnosis with the histological parameters of the distal esophagus determined using the EoE-HSS score criteria. Red symbol represents PPI-R EoE and blue symbol PPI-UR EoE. (B) DEGs and associated pathways that correlated with peak eosinophil count, DIS, BZH, and total histological score in PPI-UR EoE at diagnosis.
Common and unique transcriptome network and pathways between PPI-R EoE and PPI-UR EoE at diagnosis. To determine the common and unique expressed genes between PPI-R EoE and PPI-UR EoE at diagnosis we mapped the DEGs at diagnosis in PPI-R EoE onto the PPI-UR EoE (Supplemental Table 10). We identified a total of 1,889 common DEGs between PPI-R EoE and PPI-UR EoE at diagnosis and 407 unique DEGs in PPI-R EoE and 2,195 DEGs in PPI-UR EoE (Figure 4A and Supplemental Table 10, A–C). GO analysis of unique DEGs in PPI-R EoE identified enrichment of pathways associated with “hydrogen peroxide catabolic process,” “cellular oxidant detoxification,” “triglyceride homeostasis,” “xenobiotic metabolic process,” “positive regulation of nitric-oxide synthase biosynthetic process,” and “response to zinc ion and oxygen transport” (Figure 4B and Supplemental Table 10D). K-mean clustering interaction network analysis performed on the DEGs identified in the GO analysis revealed strong network interactions between genes, including CYP2E1, HNF4A, APOA3, APOC4 and MLXIPL, and BGLAP. Other DEGs, including PLN, MB, HBB, HBA2, SLC30A10, and TPO, demonstrated low level interactivity (Figure 4C). The 2,195 unique DEGs in PPI-UR EoE were enriched for pathways involved in “sister chromatid cohesion,” “cell division,” “mitotic nuclear division,” “DNA replication initiation,” “DNA replication,” “chromosome segregation,” “CENP-A containing nucleosome assembly,” “DNA strand elongation involved in DNA replication,” “anaphase-promoting complex- dependent catabolic process,” and “DNA replication checkpoint” (Figure 4B and Supplemental Table 10E). K-mean clustering network analysis of DEGs in PPI-UR EoE identified 3 major interaction clusters associated with cell cycle, cell division, and proliferation (Figure 4D). The cluster 1 protein network consisted of genes, including RPA3, BRIP1, POLD3, RRM1, FEN1, VRK1, RPA3, MAD2L2, INHBA, BRIP1, RPA3, MAD2L2, BRIP1, POLD3, and FEN1, that are involved in “DNA replication cell cycle,” “cell processes,” and “mitotic cell cycle DNA metabolic processes” (Supplemental Table 10F). Cluster 2 consisted of genes, including PSMA4, CDC6, PSMA3, PSME2, FBXO5, MISP, POLE2, and TIPIN that are involved in “regulation of cell cycle phase transition,” “mitotic cell cycle process,” “regulation of cell cycle process,” and “regulation of mitotic cell cycle and cell cycle process.” Cluster 3 consisted of genes, including AURKA, NCAPH, KNSTRN, NCAPG, HAUS8, NCAPH, KNSTRN, NCAPG, CCNB1, and NDC80, involved in “chromosome segregation,” “cell division,” “cell cycle,” “mitotic cell cycle,” “cell cycle process,” and “chromosome segregation” (Figure 4D and Supplemental Table 10, G and H). Collectively, these studies reveal a highly enriched esophageal epithelial proliferative transcriptome unique to PPI-UR EoE at diagnosis.
 Figure 4
Figure 4Common and unique transcriptome network and pathways between PPI-R and PPI-UR EoE at diagnosis. (A) Venn diagram depicting the common and unique DEGs between individuals acting as healthy controls and PPI-R patients and individuals acting as healthy controls and PPI-UR patients at diagnosis. (B) Gene ontology enrichment analysis of unique DEGs in PPI-R and PPI-UR patients at diagnosis. (C) K-means clustering interaction network on the DEGs associated with top enriched biological processes in PPI-R EoE at diagnosis and (D) K-means clustering interaction networks (clusters 1–3) on the DEGs associated with top enriched biological processes in PPI-UR EoE at diagnosis.
PPI effect on the transcriptome profiles between different disease groups. Upon diagnosis, individuals with EoE underwent 8- to 12-week PPI therapy, and follow-up endoscopic, histologic, and transcriptomic analyses were performed. In PPI-R individuals, we observed that PPI therapy induced disease remission characterized by a significant reduction in histologic (eos/HPF, BZH, and DIS) and endoscopic involvement (EFRES) (Table 2 and Supplemental Table 1). Transcriptomic analyses revealed that PPI therapy in PPI-R individuals with EoE led to a significant reduction in DEGs compared with that in individuals acting as controls (2,296 vs. 979 DEGs, Padj < 0.05, FC > 2; Figure 5 and Supplemental Table 11). Mapping the DEGs in PPI-R individuals with EoE at diagnosis onto the DEGs following PPI therapy revealed 1,649 genes that are PPI responsive, 647 that are PPI unresponsive, and 332 that are PPI-induced genes in PPI-R EoE (Padj < 0.05, FC > 2, Figure 5A and Supplemental Table 11A). Notably, differentially expressed hematopoietic cell–specific genes for eos, mast cells, and CD4+ Th2 cells returned to levels comparable to those of control individuals (Supplemental Figure 3A). Furthermore, the dedifferentiated esophageal epithelial transcriptomic signature observed at diagnosis resembled that of control individuals with enrichment of a mature differentiated esophageal epithelial signature (Supplemental Figure 3B). Consistent with the observed reduction in inflammatory and histological alterations, the genes that were elevated in PPI-R EoE at diagnosis associated with the biological processes “immune response” and “inflammatory response” resembled that of control individuals (Supplemental Figure 4A). Furthermore, the DEGs upregulated at diagnosis associated with the biological processes “cellular response to LPS,” “defense response to virus,” “IFN-gamma-mediated signaling,” and “type I IFN signaling pathways,” and downregulated DEGs involved in “cellular response to zinc ion,” “triglyceride homeostasis,” and “xenobiotic metabolic process pathways” also returned to levels comparable to control individuals (Supplemental Figure 4B and Supplemental Table 11, A and C). Notably, we identified n = 647 DEGs that were PPI unresponsive in PPI-R individuals with EoE (Figure 5A and Supplemental Table 11D). The DEGs that were PPI unresponsive were enriched for genes involved in the biological processes, including “arachidonic acid metabolic process” (GPX1, CYP2D7, CYP2D6, PLA2G4B, ALOX15, ALOX12, CYP4F12), “Rap1/Ras signaling pathway” (PDGFRB, RASA4B, CALML6, PLA2G4B, FLT4, CALML4, PLA2G6, VEGFA, FGF17, RASA4, JMJD7, PLA2G4B, PDGFD, GNB3, LAT, FGF22), and “neutrophil extracellular trap formation” (H4C8, H2BC9, H2AW, HDAC10, ITGA2B, H2AC19, CYBA, AGER, SEL, H2AC20, CTSG, H4C4, H4C5) (Supplemental Table 11, A and D).
 Figure 5
Figure 5The effect of PPI treatment on the transcriptome profiles between PPI-R and PPI-UR EoE. (A and B) Venn diagram of the DEGs at diagnosis versus after PPI treatment in PPI-R (A) and PPI-UR (B) EoE (P < 0.05, FC > 2) (C) Venn diagram of disease-associated DEGs in PPI-R EoE and disease-associated DEGs in PPI-UR EoE. (D) Biological processes associated with disease-associated DEGs in PPI-R EoE, and (E) biological processes associated with disease-associated DEGs in PPI-UR EoE. (F) Biological processes that are common disease drivers associated with EoE.
Finally, there were n = 332 DEGs induced by PPI therapy in PPI-R individuals with EoE that were enriched for “gated channel activity” (KCND1, GRIN3B, TRPA1, KCNH4, CACNA1C, CACNG8, KCNE4, ASIC3, CNGB3, CACNB2, CACNA1H, ZACN, TMC3, HCN3, LRRC26, GRIK3, CNGA4, GABRR2) (Figure 5A and Supplemental Table 11, A and E).
In PPI-UR individuals with EoE, 8- to 12-week PPI therapy did not demonstrate any statistically significant reduction in eos/HPF (peak eos/HPF, 87.80 ± 46.9 vs. 132.2 ± 48.7, at diagnosis vs. following PPI trial; n = 5; mean ± SD) or reduction in histologic or endoscopic disease involvement (Table 2, Supplemental Table 1, and Supplemental Figure 1). While PPI trial in PPI-UR individuals with EoE did not induce disease remission (Table 2), transcriptomic analyses revealed that PPI trial reduced the number of DEGs when compared with individuals acting as controls (4,084 vs. 3283 DEGs, P < 0.05, FC > 2, Figure 5B). Mapping the DEGs in PPI-UR individuals with EoE at diagnosis onto the DEGs following PPI therapy revealed 1,286 genes that are PPI responsive, 2,798 that are PPI unresponsive, and 485 that are PPI-induced genes in PPI-UR EoE (P < 0.05, FC > 2, Figure 5B and Supplemental Table 11B). Consistent with the persistent inflammation and esophageal epithelial remodeling, the differentially expressed hematopoietic cell–specific genes for eos, mast cells and CD4+ Th2 cells remained significantly dysregulated, particularly the mast cell DEG signature (Supplemental Figure 3A). Furthermore, the dedifferentiated esophageal epithelial transcriptomic signature persisted in PPI-UR individuals with EoE following PPI therapy (Supplemental Figure 3B and Supplemental Table 11, B, F, and G). The level of expression of DEGs in PPI-UR EoE at diagnosis associated with the biological processes “immune response” and “inflammatory response” was reduced following PPI therapy but, in comparison to PPI-UR EoE at diagnosis, remained differentially expressed as compared with that of individuals acting as controls (Supplemental Figure 4A). Intriguingly, the common genes associated with the enriched biological processes in PPI-R EoE, such as “cellular response to LPS,” “defense response to virus,” and “type I IFN pathway,” and the genes and biological pathways specifically enriched in PPI-UR EoE at diagnosis, including “positive regulation of interleukin-6 production” and “sister chromatid cohesion,” remained differentially expressed following PPI therapy (Supplemental Figure 4, B and C). GO analysis revealed that the 1,286 DEGs that were PPI responsive in PPI-UR EoE were enriched for genes involved in “B cell receptor signaling pathway,” “immunoglobulin receptor binding,” “immunoglobulin complex,” and “complement activation, classical pathway” (Supplemental Table 11G). Paired analyses comparing gene expression between PPI-UR EoE at diagnosis and following PPI therapy identified no DEGs at a cutoff threshold of Padj < 0.05, FC > 2.
Disease-specific gene signature in PPI-R and PPI-UR EoE. Our combined clinical and transcriptomic analyses suggest that the n = 1,649 DEGs that were PPI responsive in PPI-R EoE and the n = 2,798 DEGs that were PPI unresponsive in PPI-UR EoE are disease-specific gene drivers. To determine whether the transcriptomic programs that drive PPI-R and PPI-UR EoE are similar, we mapped the n = 1,649 disease-associated DEGs in PPI-R EoE onto the n = 2,798 disease- associated DEGs in PPI-UR EoE (Figure 5C and Supplemental Table 12, A–D). We identified 509 genes that were PPI-R EoE–specific gene drivers that were enriched for the biological processes involved in “cilium movement” (CCR1, UCN, CERS6, CAMK1D, CSF1), “cholesterol biosynthetic process” (CCR1, EDN2, RTN4RL2, LILRB2, TRPV1), “neuropeptide signaling pathway” (ROBO4, EDN2, GPR15, EPAS1, ERAP1), “triglyceride homeostasis” (IL33, UNC93B1, IFIT5, EIF2AK2, IFIT1), “inflammatory response” (POMC, UCN, GALR3, LTB3R2, NXPH4), and “angiogenesis” (POMC, GREM1, CCR1, C1QA, SH2D1A) (Figure 5, C and D, and Supplemental Table 12C). There were 1,658 genes that were PPI-UR EoE–specific gene drivers, including genes enriched for “mitotic spindle assembly checkpoint” (ZWILCH, BUB1B, TTK, HASPIN, NDC80), “chromosome segregation” (TOP3B, CENPT, CDT1, CENPW, SPAG5), “mitotic cell cycle” (CDCA5, BUB1B, KIF11, SKA3, AURKA), “cell division” (ZWILCH, NCAPG2, BUB1B, KIF11, SPOUT1), “xenobiotic metabolic process” (ABCC4, ABCC1, UGT1A1, RORC, AHR), and “mitotic spindle organization” (STIL, TTK, TUBG2, KIF11, AURKC) (Figure 5, C and E, and Supplemental Table 12D). Importantly, these analyses also revealed n = 1,140 genes that we defined as common EoE disease gene drivers, which included “inflammatory response” (GMFB, GMFG, RASL11A, IL5RA), “immune response” (TPO2A, CD40, ZNF493, IL26, IHH), “defense response to virus” (STAT1, STING1, IRF1, TLR8, TLR3, TLR2), “detoxification of copper ion” (MT2A, MT1A, MT1L, MT1M, MT1F) “cellular zinc ion homeostasis” (MT2A, MT1A, MT1L, MT1M, MT1F, MT1G), “defense response to virus” (IFITM3, RTP4, CD40, DDX60L, IFIT3), and “chemotaxis” (CCL13, CCL23, CXCL8, PTGDR2, PLAUR) (Figure 5F and Supplemental Table 12B) genes. Collectively, these studies indicate the presence of common and unique transcriptomic elements that underlie PPI-R and PPI-UR EoE disease phenotypes.









