Cell-associated HIV-1 RNA predicts viral rebound and disease progression after discontinuation of temporary early ART
Alexander O. Pasternak,1 Marlous L. Grijsen,2,3 Ferdinand W. Wit,2,4,5,6 Margreet Bakker,1 Suzanne Jurriaans,7 Jan M. Prins,2 and Ben Berkhout1
1Laboratory of Experimental Virology, Department of Medical Microbiology, and
2Department of Internal Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
4Global Health program, Amsterdam Public Health research institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
5Amsterdam Institute for Global Health and Development, Amsterdam, Netherlands.
6HIV Monitoring Foundation, Amsterdam, Netherlands.
7Laboratory of Clinical Virology, Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
Address correspondence to: Alexander O. Pasternak, AMC, Room K3-113B, Meibergdreef 15, 1105 AZ Amsterdam, Netherlands. Phone: 31.20.5668572; Email: a.o.pasternak@amsterdamumc.nl.
Find articles by
Pasternak, A.
in:
PubMed
|
Google Scholar
|

1Laboratory of Experimental Virology, Department of Medical Microbiology, and
2Department of Internal Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
4Global Health program, Amsterdam Public Health research institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
5Amsterdam Institute for Global Health and Development, Amsterdam, Netherlands.
6HIV Monitoring Foundation, Amsterdam, Netherlands.
7Laboratory of Clinical Virology, Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
Address correspondence to: Alexander O. Pasternak, AMC, Room K3-113B, Meibergdreef 15, 1105 AZ Amsterdam, Netherlands. Phone: 31.20.5668572; Email: a.o.pasternak@amsterdamumc.nl.
Find articles by Grijsen, M. in: PubMed | Google Scholar
1Laboratory of Experimental Virology, Department of Medical Microbiology, and
2Department of Internal Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
4Global Health program, Amsterdam Public Health research institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
5Amsterdam Institute for Global Health and Development, Amsterdam, Netherlands.
6HIV Monitoring Foundation, Amsterdam, Netherlands.
7Laboratory of Clinical Virology, Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
Address correspondence to: Alexander O. Pasternak, AMC, Room K3-113B, Meibergdreef 15, 1105 AZ Amsterdam, Netherlands. Phone: 31.20.5668572; Email: a.o.pasternak@amsterdamumc.nl.
Find articles by Wit, F. in: PubMed | Google Scholar
1Laboratory of Experimental Virology, Department of Medical Microbiology, and
2Department of Internal Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
4Global Health program, Amsterdam Public Health research institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
5Amsterdam Institute for Global Health and Development, Amsterdam, Netherlands.
6HIV Monitoring Foundation, Amsterdam, Netherlands.
7Laboratory of Clinical Virology, Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
Address correspondence to: Alexander O. Pasternak, AMC, Room K3-113B, Meibergdreef 15, 1105 AZ Amsterdam, Netherlands. Phone: 31.20.5668572; Email: a.o.pasternak@amsterdamumc.nl.
Find articles by Bakker, M. in: PubMed | Google Scholar
1Laboratory of Experimental Virology, Department of Medical Microbiology, and
2Department of Internal Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
4Global Health program, Amsterdam Public Health research institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
5Amsterdam Institute for Global Health and Development, Amsterdam, Netherlands.
6HIV Monitoring Foundation, Amsterdam, Netherlands.
7Laboratory of Clinical Virology, Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
Address correspondence to: Alexander O. Pasternak, AMC, Room K3-113B, Meibergdreef 15, 1105 AZ Amsterdam, Netherlands. Phone: 31.20.5668572; Email: a.o.pasternak@amsterdamumc.nl.
Find articles by Jurriaans, S. in: PubMed | Google Scholar
1Laboratory of Experimental Virology, Department of Medical Microbiology, and
2Department of Internal Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
4Global Health program, Amsterdam Public Health research institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
5Amsterdam Institute for Global Health and Development, Amsterdam, Netherlands.
6HIV Monitoring Foundation, Amsterdam, Netherlands.
7Laboratory of Clinical Virology, Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
Address correspondence to: Alexander O. Pasternak, AMC, Room K3-113B, Meibergdreef 15, 1105 AZ Amsterdam, Netherlands. Phone: 31.20.5668572; Email: a.o.pasternak@amsterdamumc.nl.
Find articles by Prins, J. in: PubMed | Google Scholar
1Laboratory of Experimental Virology, Department of Medical Microbiology, and
2Department of Internal Medicine, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
3Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
4Global Health program, Amsterdam Public Health research institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
5Amsterdam Institute for Global Health and Development, Amsterdam, Netherlands.
6HIV Monitoring Foundation, Amsterdam, Netherlands.
7Laboratory of Clinical Virology, Department of Medical Microbiology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands.
Address correspondence to: Alexander O. Pasternak, AMC, Room K3-113B, Meibergdreef 15, 1105 AZ Amsterdam, Netherlands. Phone: 31.20.5668572; Email: a.o.pasternak@amsterdamumc.nl.
Find articles by Berkhout, B. in: PubMed | Google Scholar
Published February 25, 2020 - More info
JCI Insight. 2020;5(6):e134196. https://doi.org/10.1172/jci.insight.134196.
© 2020 American Society for Clinical Investigation
Received: October 14, 2019; Accepted: February 19, 2020
-
Results
Study participants and variables. All 64 participants of the Primo-SHM trial who completed the trial in the Academic Medical Center of the University of Amsterdam (AMC) were included in this study. Baseline and treatment characteristics of the study participants are shown in Table 1. In brief, the median age was 39.8 years and 95.3% were males. Most participants (82.8%) were MSM, and 84.4% were infected with HIV-1 subtype B. The study design is shown in Figure 1. Participants received 60 weeks’ (n = 29), 24 weeks’ (n = 23), or no (n = 12) early ART.
 Figure 1
Figure 1Schematic of the study. Participants received either 60 weeks’ (n = 29), 24 weeks’ (n = 23), or no (n = 12) early ART. Variables were measured at 3 time points (shown in red): baseline (corresponding to the start of early ART for the 60-week and 24-week arms), early ART interruption (for 60-week and 24-week arms), and virological set point (36 weeks from early ART interruption or baseline). Numbers of participants included in the corresponding analyses: baseline (n = 44), treatment interruption (n = 51), and set point (n = 56).
All virological and immunological variables — plasma VL, CA HIV-1 unspliced (US) (gag) and multiply spliced (MS) (tat/rev) RNA, total HIV-1 DNA, CD4+ count, and CD4/CD8 ratio — were measured at baseline (corresponding to the start of early ART for the 60-week and 24-week arms) and at the virological set point. The latter was defined, in accordance with the original Primo-SHM trial design (28), at 36 weeks from early ART interruption (TI) or, for the no-treatment arm, at 36 weeks from baseline, to allow for stabilization of the plasma VL in untreated and treated patients (29, 30) and to minimize dropout due to rapid disease progression (Figure 1). During early ART, plasma VL, CD4+ count, and CD4/CD8 ratio were measured weekly during the first month, monthly during the second and third months, and every 3 months for the remainder of the period on therapy. After TI, these variables were measured bimonthly for the first month, monthly during the second and third months, and subsequently every 3 months. US and MS RNA and total DNA were measured every 12 weeks throughout the early ART periods. For the present study, measurements of these variables on the latest available visit before TI were included in the analysis as candidate predictors of viral rebound and CD4+ cell loss upon TI. Because MS RNA was rarely detectable during ART, participants were stratified into those with detectable or undetectable MS RNA in any on-ART measurement, and MS RNA detectability on ART was included in this analysis as a categorical variable.
As expected, levels of all virological markers decreased on ART and increased again after TI, while the opposite dynamics were observed for the immunological markers (CD4+ count and CD4/CD8 ratio) (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.134196DS1). Detailed analysis of the longitudinal dynamics of virological and immunological markers on early ART, as well as comparisons between baseline and post-TI set point values of the markers per treatment arm will be reported separately. At baseline and at set point, all variables significantly correlated between each other: positive correlations were observed in any pair of virological markers, as well as between CD4+ count and CD4/CD8 ratio, whereas virological markers negatively correlated with immunological markers (Figure 2, A and C). At TI, we only observed (positive) correlations between CD4+ count and CD4/CD8 ratio and between US RNA and total DNA, whereas no significant correlations were detected between virological and immunological markers (Figure 2B). Strong positive correlations were also observed for all virological and immunological markers between the time points (Figure 2D). For each of these 3 time points, the measured variables were assessed as candidate predictors of the corresponding clinical endpoints, as follows: baseline variables were assessed for the prediction of response to ART, TI variables for the prediction of viral rebound and disease progression, and set point variables for the prediction of disease progression. To compare the predictive power of different explanatory variables for the corresponding clinical endpoints, only participants without missing values on any variable (per time point) were included in the corresponding analyses (Figure 1). In addition, a number of demographic and clinical covariates, such as age, sex, HIV-1 transmission route, HIV-1 subtype, Fiebig stage of early infection, early ART duration, NRTI backbone, and ART class, were included in the analyses as appropriate.
 Figure 2
Figure 2Correlation matrices of clinical and virological variables. (A–C) Correlations between different variables measured at (A) baseline (BL), (B) early ART interruption (TI), and (C) virological set point (SP). Participants from all 3 arms were included in the BL and SP correlation analyses; participants from 24- and 60-week treatment arms were included in the TI correlation analysis. Numbers of measurements are shown for every variable and for every time point. (D) Correlations between different time points per variable. Heatmaps indicate the strength of positive (blue) or negative (red) correlations. Spearman’s rho coefficients are depicted below the diagonal and P values are above the diagonal. US RNA, unspliced RNA; MS RNA, multiply spliced RNA.
CA HIV-1 RNA predicts virological and immunological response to ART. First, we assessed the predictive value of baseline variables for the time to achieve virological suppression (plasma VL < 50 copies/mL) on early ART. All participants included in the baseline analysis, except 1, achieved virological suppression on therapy within a median of 115 (IQR, 84–168) days, counting from the baseline to the first undetectable plasma VL measurement. The single participant who did not achieve virological suppression was censored from the analysis at TI. For the univariate Kaplan-Meier analysis, levels of every potential continuous predictor were stratified into “high” and “low” strata based on median values, and times to virological suppression were compared between participants with high and low values of every predictor. In this analysis, among all potential predictors, only plasma VL, US RNA, and total DNA were significantly associated with virological suppression (P = 0.0029, P = 0.0035, and P = 0.0007, respectively) (Figure 3 and Supplemental Table 1). Univariable Cox analysis, in which all continuous variables were included without categorization, revealed that the same variables, as well as CD4/CD8 ratio, were significantly associated with virological suppression (Table 2). However, only CD4/CD8 ratio remained a significant independent predictor in the final multivariable Cox regression model (aHR = 1.14 [1.01–1.30], and P = 0.035). In addition, a trend toward US RNA predicting virological suppression was observed (aHR = 0.72 [0.51–1.02], and P = 0.063) (Table 2).
 Figure 3
Figure 3Pre-ART predictors of time to virological suppression on early ART. Kaplan-Meier survival analysis for plasma VL, CA HIV US RNA, CA HIV MS RNA, total HIV DNA (VD), CD4+ count, and CD4/CD8 ratio, measured at the start of early ART (n = 44). The endpoint of the analysis was virological suppression less than 50 copies/mL. Levels of every potential predictor were stratified into “high” and “low” strata based on median values. HRs and 95% CIs were calculated using Mantel-Haenszel method. P values were calculated using log-rank tests.
 Table 2
Table 2Baseline variables associated with time to achieve virological suppression (plasma VL < 50 copies/mL) on early ART (n = 44; Cox proportional hazards analysis)
We then asked whether any of the baseline variables would be predictive for virological suppression at an early time point after ART initiation. Levels of every potential predictor were compared between participants who did and did not achieve virological suppression at 12 weeks of ART by Mann-Whitney U or Fisher’s tests. In this analysis, only US RNA was significantly associated with virological suppression (P = 0.032), whereas plasma VL was not predictive (Figure 4 and Supplemental Table 2). The same result was obtained by univariable and multivariable logistic regression analyses (adjusted odds ratio [aOR] = 0.48 [0.25–0.93], P = 0.029) (Table 3).
 Figure 4
Figure 4Pre-ART predictors of virological suppression at 12 weeks of early ART. Plasma VL, CA HIV US and MS RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio, measured at the start of early ART, were compared between participants with undetectable (n = 19, blue dots) and detectable (n = 24, red dots) plasma VL at 12 weeks of early ART. P values were calculated using Mann-Whitney U tests. Open circles depict undetectable values, censored to 50% of assay detection limits.
 Table 3
Table 3Baseline variables associated with virological suppression (plasma VL < 50 copies/mL) at 12 weeks of early ART (n = 43; logistic regression analysis)
Having shown the predictive value of CA RNA for virological response to early ART, we next investigated whether it would also be predictive for the immunological response to therapy. Normalization of CD4/CD8 ratio to more than 1 is considered an important measure of immunological response to ART, and a low CD4/CD8 ratio is a prognostic marker for both opportunistic infections and non-AIDS morbidity and mortality (31, 32). Because many participants, especially those who were treated for 24 weeks, did not achieve a CD4/CD8 ratio of more than 1 by the time of TI, we did not consider survival analysis appropriate because all these participants would have been censored, which would have biased the results. Instead, we assessed the predictive power of the baseline variables for the normalization of CD4/CD8 ratio in the first year of early ART (only the 60-week treatment arm was included in this analysis). We compared levels of every potential predictor between participants who did and did not achieve a CD4/CD8 ratio of more than 1 at 48 weeks of ART. In addition to the baseline CD4/CD8 ratio itself (P = 0.039), only MS RNA was significantly associated with immunological response (P = 0.0043) (Figure 5 and Supplemental Table 3). In the univariable and multivariable logistic regression analyses, only MS RNA was significantly predictive of immunological response to ART (aHR = 0.14 [0.03–0.73], P = 0.020), whereas baseline CD4/CD8 ratio was not predictive (Table 4).
 Figure 5
Figure 5Pre-ART predictors of CD4/CD8 ratio normalization at 48 weeks of early ART. Plasma VL, CA HIV US and MS RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio, measured at the start of early ART, were compared between participants with CD4/CD8 ratio less than 1 (n = 8, blue dots) and more than 1 (n = 16, red dots) at 48 weeks of early ART. P values were calculated using Mann-Whitney U tests. Open circles depict undetectable values, censored to 50% of assay detection limits.
 Table 4
Table 4Baseline variables associated with CD4/CD8 ratio more than 1 at 48 weeks of early ART (n = 24; logistic regression analysis)
CA HIV-1 RNA predicts the magnitude of viral rebound after TI. All individuals who interrupted early ART (n = 51) achieved viral rebound to more than 50 copies/mL within 252 days and to more than 400 copies/mL within 574 days, counting from the moment of TI to the first plasma VL measurement higher than the corresponding threshold (Supplemental Figure 2). The median magnitude of viral rebound, measured as plasma VL at the virological set point (36 weeks after TI) was 4.43 (IQR, 3.63–4.94) log10 copies/mL. We first assessed the predictive power of variables measured before TI for the magnitude of viral rebound. In both uni- and multivariable analysis, US RNA was the only variable significantly associated with plasma VL at set point (P = 0.003, and P = 0.020, respectively) (Figure 6A). We reasoned, however, that this association might be partly explained by the predictive power of pre-ART virological markers for viral rebound. Although pre-ART CD4+ count, CD4/CD8 ratio, US RNA, and plasma VL were all significantly associated with viral rebound in the univariable model, only US RNA remained as an independent predictor in the multivariable model (P = 0.001). In addition, pre-ART total HIV-1 DNA, which was not associated with viral rebound in the univariable model, became strongly negatively associated in the multivariable model (P = 2.9 × 10–4) (Figure 6B). Because the latter 2 pre-ART variables were strongly positively correlated (rho = 0.65, P = 7.7 × 10–8) (Figure 2A), the apparent lack of predictive power of total DNA for viral rebound in the univariable model can be explained by masking of this effect by the high positive predictive power of US RNA. Controlling for US RNA in the multivariable model revealed the negative predictive power of total DNA.
 Figure 6
Figure 6Predictors of magnitude of viral rebound after interruption of early ART. (A and B) Coefficient plots of variables measured (A) at early ART interruption (n = 51) or (B and C) at the start of early ART (baseline) (n = 43). Effect sizes and 95% CIs for the associations of the variables with plasma VL at the virological set point were calculated by fitting univariable and multivariable generalized linear models. For baseline, 2 models are shown, including either US RNA and total HIV-1 DNA separately (model I, B) or the US RNA/total DNA ratio as a single variable (model II, C). US RNA/total DNA ratio was calculated taking into account that 106 PBMCs contain 1 μg of total RNA (74). (D) The final multivariable model. Units of measurement of total HIV-1 DNA are log10 copies/106 PBMCs and of US are log10 copies/μg total RNA. Significant associations are marked in red.
Given the opposite associations of pre-ART US RNA and total DNA with viral rebound, we attempted to further improve the multivariable model by introducing the US RNA/total DNA ratio into the model as a single variable (Figure 6C). Indeed, the association of the pre-ART US RNA/total DNA ratio with viral rebound was stronger than that of either US RNA or total DNA alone, and its inclusion in the multivariable model resulted in an improved model fit (Akaike Information Criterion, 97.8 vs. 102.8). In this improved model, only the US RNA/total DNA ratio was independently predictive of viral rebound (P = 7.5 × 10–5). Pre-ART and TI variables that were predictive of viral rebound were subsequently selected for the final multivariable model (Figure 6D). In this analysis, the associations of TI US RNA and pre-ART US RNA/total DNA ratio with the magnitude of viral rebound remained significant (B = 0.35 [95% CI = 0.09–0.60], P = 0.007; and B = 0.62 [0.37–0.87], P = 1 × 10–6, respectively).
CA HIV-1 RNA predicts the time to viral rebound and CD4+ T cell decline after TI. Median times to viral rebound of more than 50 and more than 400 copies/mL were 35 (IQR, 28–56) days and 55 (IQR, 28–60) days, respectively (Supplemental Figure 2). Because no pre-ART virological marker was predictive for time to viral rebound after TI (Supplemental Table 4), we assessed the predictive power of variables measured before TI for the time to viral rebound. Among the clinical and virological variables, only high US RNA level was significantly associated with shorter time to viral rebound of both more than 50 and more than 400 copies/mL (HR = 0.41 [0.21–0.78], P = 0.0070; and HR = 0.36 [0.18–0.70], P = 0.0026, respectively) (Figure 7). This was confirmed in a subset of participants with plasma VL less than 50 copies/mL at the measurement time points (n = 44) (Supplemental Figure 3). In addition, younger age was associated with longer time to viral rebound of more than 50 but not of more than 400 copies/mL (P = 0.020), and HIV-1 subtype B was marginally associated with shorter time to viral rebound of both more than 50 and more than 400 copies/mL (P = 0.062, and P = 0.059, respectively) (Supplemental Table 5). Surprisingly, longer time on ART (60 vs. 24 weeks) was not associated with longer time to viral rebound, and a trend in the opposite direction was observed, as the 24-week arm demonstrated longer times to viral rebound of more than 400 copies/mL (P = 0.10). The predictive power of TI US RNA was confirmed in the univariable and multivariable Cox analyses because it was the only variable significantly associated with time to viral rebound of both more than 50 and more than 400 copies/mL (aHR = 1.47 [1.02–2.11], P = 0.040; and aHR = 1.47 [1.00–2.14], P = 0.047) (Table 5).
 Figure 7
Figure 7On-ART predictors of time to viral rebound after interruption of early ART. Kaplan-Meier survival analysis for CA HIV US RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio, measured at the latest time point available before interruption of early ART, as well as detectability of CA HIV MS RNA at any time point on early ART, and time on early ART (60 or 24 weeks) (n = 51). The endpoint of the analysis was viral rebound to more than 50 or more than 400 copies/mL. Levels of US RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio were stratified into “high” and “low” strata based on median values. HRs and 95% CIs were calculated using Mantel-Haenszel method. P values were calculated using log-rank tests. det, detectable; ud, undetectable.
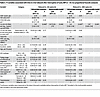 Table 5
Table 5TI variables associated with time to viral rebound after interruption of early ART (n = 51; Cox proportional hazards analysis)
We next sought to establish the predictive role of variables measured before TI for subsequent disease progression. The latter was protocol-defined as the time to experience a drop in the CD4+ count below 350 cells/mm3 or to restart ART. All except 3 participants achieved this composite endpoint by the end of the observation period within a median of 555 (IQR, 102–1350) days from TI. These 3 participants were censored from the analysis at the end of the observation period. Of the remaining 48 participants, 35 reached the CD4+ count of less than 350 cells/mm3, and 13 restarted ART with higher CD4+ counts (by the trial protocol, participants had to start or restart ART in the case of 2 consecutive CD4+ counts less than 350 cells/mm3, symptomatic HIV-1 disease, or physician or participant insistence on starting or restarting ART). In addition to high CD4+ count (HR = 3.12 [1.64–5.93], P = 0.0005), undetectable MS RNA on ART (HR = 0.16 [0.05–0.53], P = 0.0027) and low total HIV-1 DNA at TI (HR = 0.46 [0.25–0.85], P = 0.014) were significantly associated with longer disease progression, while low US RNA was marginally significant (HR = 0.55 [0.30–1.00], P = 0.052) (Figure 8). In addition, HIV-1 subtype B was associated with shorter disease progression (HR = 2.47 [1.29–4.72], P = 0.0062) (Supplemental Table 6). Again, time on ART (60 vs. 24 weeks) was not significantly associated with disease progression. These associations were supported by the univariable Cox regression analysis (Table 6), but only CD4+ count and US RNA remained as independent predictors in the final multivariable Cox model (aHR = 0.65 [0.55–0.77], P = 7.4 × 10–7; and aHR = 1.59 [1.11–1.28], P = 0.012, respectively).
 Figure 8
Figure 8On-ART predictors of disease progression after interruption of early ART. Kaplan-Meier survival analysis for CA HIV US RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio, measured at the latest time point available before interruption of early ART, as well as detectability of CA HIV MS RNA at any time point on early ART, and time on early ART (60 or 24 weeks) (n = 51). The composite endpoint of the analysis was either a CD4+ count measurement of less than 350 cells/mm3 or restart of ART. Levels of US RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio were stratified into “high” and “low” strata based on median values. HRs and 95% CIs were calculated using Mantel-Haenszel method. P values were calculated using log-rank tests. det, detectable; ud, undetectable.
 Table 6
Table 6TI variables associated with disease progression (CD4+ count < 350 cells/mm3 or restart ART) after interruption of early ART (n = 51; Cox proportional hazards analysis)
CA HIV-1 RNA predicts disease progression in the absence of treatment. Finally, we sought to establish the predictive role of variables measured in untreated infection (at the virological set point) for the subsequent disease progression (as defined above). Participants from all 3 arms, including those who were randomized to no treatment during early ART, were included in this analysis. All virological and clinical variables measured at the set point were significantly predictive of disease progression, with high MS RNA level, low CD4+ count, and low CD4/CD8 ratio demonstrating particularly strong predictive power with hazard ratios of 0.22 (0.11–0.44), 14.48 (6.58–31.85), and 8.87 (4.20–18.75), respectively, and P value less than 0.0001 for all 3 predictors (Figure 9). Having been treated with temporary early ART was also significantly associated with longer disease progression (HR = 0.19 [0.06–0.61], P = 0.0047), confirming the earlier results of Grijsen et al. (28). HIV-1 subtype B was marginally associated with faster disease progression (P = 0.053) (Supplemental Table 7). These associations were confirmed by the univariable Cox regression analysis because all virological and clinical variables remained significantly associated with disease progression (Table 7). However, only CD4+ count and MS RNA were retained as independent predictors in the final multivariable Cox model (aHR = 0.55 [0.42–0.72], P = 1.1 × 10–5; and aHR = 1.46 [1.07–2.01], P = 0.019, respectively). The same results were obtained when the drop of CD4+ count below 350 cells/mm3 was used as the sole endpoint, censoring the participants who (re)started ART with higher CD4+ counts (data not shown).
 Figure 9
Figure 9Predictors of disease progression in the absence of treatment. Kaplan-Meier survival analysis for plasma VL, CA HIV US and MS RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio, measured at the virological set point (36 weeks after interruption of early ART or randomization), as well as having been treated with early ART or not (n = 56). The composite endpoint of the analysis was either a CD4+ count measurement of less than 350 cells/mm3 or (re)start of ART. Levels of plasma VL, US RNA, MS RNA, total HIV DNA, CD4+ count, and CD4/CD8 ratio were stratified into “high” and “low” strata based on median values. HRs and 95% CIs were calculated using Mantel-Haenszel method. P values were calculated using log-rank tests.
 Table 7
Table 7Set point variables associated with the rates of disease progression (time to CD4+ count < 350 cells/mm3 or restart ART) after virological set point (n = 56; Cox proportional hazards analysis)


















