Research ArticleCell biologyGastroenterologyMuscle biology
Open Access | ![]() 10.1172/jci.insight.188669
10.1172/jci.insight.188669
Rapid cyclic stretching of cultured human visceral smooth muscle cells promotes a synthetic, proinflammatory phenotype
Sharon M. Wolfson,1,2 Katherine Beigel,3 Sierra E. Anderson,1 Brooke Deal,1 Molly Weiner,1,4 Se-Hwan Lee,5 Deanne M. Taylor,1,2,3 Su Chin Heo,4,5,6,7,8 Robert O. Heuckeroth,1,2 and Sohaib K. Hashmi1,4,6,9
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by Wolfson, S. in: PubMed | Google Scholar
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by
Beigel, K.
in:
PubMed
|
Google Scholar
|

1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by Anderson, S. in: PubMed | Google Scholar
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by Deal, B. in: PubMed | Google Scholar
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by Weiner, M. in: PubMed | Google Scholar
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by Lee, S. in: PubMed | Google Scholar
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by
Taylor, D.
in:
PubMed
|
Google Scholar
|
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by Heo, S. in: PubMed | Google Scholar
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by
Heuckeroth, R.
in:
PubMed
|
Google Scholar
|
1The Children’s Hospital of Philadelphia Research Institute and the Abramson Research Center, Philadelphia, Pennsylvania, USA.
2Department of Pediatrics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
3The Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA.
4Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
5McKay Orthopaedic Research Laboratory, Department of Orthopaedic Surgery, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
6Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, Philadelphia, Pennsylvania, USA.
7Center for Engineering Mechanobiology, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
8Translational Musculoskeletal Research Center, Corporal Michael J. Crescenz VA Medical Center, Philadelphia, Pennsylvania, USA.
9Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Address correspondence to: Robert O. Heuckeroth, The Children’s Hospital of Philadelphia Research Institute, Perelman School of Medicine at the University of Pennsylvania, Abramson Research Center — Suite 1116i, 3615 Civic Center Blvd., Philadelphia, Pennsylvania 19104, USA. Phone: 215.590.1209; Email: heuckerothr@chop.edu. Or to: Sohaib Hashmi, Department of Internal Medicine, Division of Gastroenterology and Hepatology, Hospital of the University of Pennsylvania, Department of Bioengineering, The University of Pennsylvania School of Engineering and Applied Science, 3400 Spruce St., Philadelphia, Pennsylvania, 19104, USA. Email: sohaib.hashmi@pennmedicine.upenn.edu.
Authorship note: SKH and ROH contributed equally to this work.
Find articles by
Hashmi, S.
in:
PubMed
|
Google Scholar
|
Published September 16, 2025 - More info
JCI Insight. 2025;10(20):e188669. https://doi.org/10.1172/jci.insight.188669.
© 2025 Wolfson et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: November 1, 2024; Accepted: September 9, 2025

-
Results
HISMCs grown on aligned scaffolds have more smooth muscle myosin heavy chain 11 (MYH11) protein and less VIM mRNA than HISMCs grown on nonaligned scaffolds. Our initial goal was to test the hypothesis that pathological mechanical stress acutely alters gene expression in contractile bowel SMC. One challenge is that SMCs cultured on hard plastic rapidly undergo phenotypic class switching from a “contractile” (MYH11-expressing) to a “synthetic” phenotype that produces extracellular matrix (ECM), migrates, and proliferates (7, 12). To study the effects of mechanical stress in a more contractile phenotype cell, we seeded HISMCs onto electrospun poly-caprolactone (PCL) scaffolds (13) coated with laminin, an ECM protein that promotes the contractile SMC phenotype (14, 15) (Figure 1, A and B). One set of PCL scaffolds was spun to have aligned fibers to promote growth of elongated spindle-shaped SMCs reported to be more contractile (16, 17). In parallel, HISMCs were cultured on laminin-coated PCL scaffolds with nonaligned fibers (Figure 1, A and B). After 72 hours with scaffolds floating freely in HISMC media, cells were fixed and stained with antibodies to MYH11, a contractile apparatus protein prominently produced in mature contractile phenotype SMCs. Pixel intensity measurements showed HISMCs grown on aligned scaffolds averaged (mean) 14% more MYH11 protein than HISMC grown on nonaligned scaffolds (aligned: 54.33 arbitrary units [AU] [32.6 AU]; nonaligned: 47.6 AU [28.28 AU], median [interquartile range]) (Mann-Whitney U test, P < 0.0001, n = 3) (Figure 1, B and C). Vimentin (VIM) mRNA, a synthetic SMC marker (18), was also less abundant in HISMCs cultured on aligned compared with nonaligned scaffolds (P = 0.0052, n = 5) (Figure 1D). In contrast, mRNA for ECM-related (COL1A1, MMP14, FN1) and contractile apparatus genes (MYH11, ACTG2, ACTA2) were statistically equivalent in HISMCs cultured on aligned versus nonaligned scaffolds (Figure 1, E–J). Based on these findings, further experiments used aligned scaffolds.
 Figure 1
Figure 1HISMC cultured on aligned nanofibrous spun scaffolds had more MYH11 protein and less VIM mRNA compared with HISMC cultured on nonaligned scaffolds. (A) Schematic of nonaligned (top) and aligned (bottom) PCL scaffolds. (B) Confocal Z-stack maximum intensity projections of HISMCs stained with antibodies to smooth muscle myosin (MYH11, magenta) and F-actin (Phalloidin-Alexa Fluor 488, green) after culture on nonaligned (top) or aligned (bottom) scaffolds for 72 hours. Scale bar: 100 μm. (C) MYH11 antibody staining was brighter in HISMCs cultured 72 hours on aligned scaffolds compared with HISMC cultured on nonaligned scaffolds (median [interquartile range] aligned: 54.33 AU [32.6 AU], nonaligned: 47.6 AU [28.28 AU], Mann-Whitney test, P < 0.0001, n = 3). (D–J) qPCR analyses for mRNA levels of smooth muscle synthetic genes (VIM, COL1A1, MMP14, FN1) showed increased VIM expression in HISMCs grown on nonaligned scaffolds (D), with similar expression of other synthetic genes. qPCR analyses demonstrated mRNA levels of smooth muscle contractile genes (MYH11, ACTG2, ACTA2) were similar for HISMCs grown on nonaligned and aligned scaffolds. VIM (mean ± SEM aligned: 0.01462 ± 0.002633, mean nonaligned: 0.025 ± 0.0002020, P = 0.0052, n = 5). ACTA2 (mean ± SEM aligned: 0.3656 ± 0.07053, mean nonaligned: 0.4986 ± 0.04450, P = 0.1494, n = 5). MYH11 (mean ± SEM aligned: 0.002982 ± 0.0006099, mean nonaligned: 0.003978 ± 0.0004698, P = 0.2322, n = 5). COL1A1 (median [interquartile range], aligned: 10.38 [12.679], nonaligned: 18.95 [4.61], P = 0.0952, n = 5). FN1 (median [interquartile range], aligned: 6.985 [6.93], nonaligned: 10.21 [8.483], P = 0.4206, n = 5). ACTG2 (mean ± SEM aligned: 0.7499 ± 0.06910, mean nonaligned: 1.009 ± 0.1477, P = 0.1249, n = 5). MMP14 (median [interquartile range], aligned: 1.16 [1.624], nonaligned: 2.319 [2.898], P = 0.2222, n = 5). **P < 0.01, ***P < 0.001.
Dynamic loading (cyclic stretching) of HISMCs leads to marked changes in gene expression. To identify early gene expression changes in response to pathologic stretch, HISMCs cultured 72 hours on aligned PCL scaffolds were subjected to cyclic uniaxial stretch (loaded) along the long axis of the cell (3% stretch, 1 Hz, 6 hours). Unloaded control scaffolds were maintained free-floating in fresh culture media for 6 hours (Figure 2A). Scaffolds were then stained (Figure 2B) or dissolved in Trizol for RNA-Seq. Bulk RNA-Seq demonstrated clear separation of loaded (stretched) versus unloaded (free-floating) HISMCs using Principal Component Analysis (PCA) (n = 4 per group; Figure 2C). Differential expression analysis using DESeq2 identified 1,239 mRNA that were more abundant (log2FC > 0.48) and 1,261 mRNA less abundant (log2FC < –0.48) in loaded compared with unloaded HISMCs (adjusted P < 0.05, DESeq2). This gene set includes markers of contractile or synthetic SMC phenotypes, inflammatory mediators, TGF-β superfamily genes, axon guidance molecules, cytoskeletal proteins, cell-cell junctional proteins, and cell-ECM interacting proteins (Figure 2D). In addition, gene set enrichment analysis (GSEA) using Hallmark gene sets from the Human Molecular Signatures Database (MSigDB) highlighted several cytokine and inflammation pathways with normalized enrichment scores (NES) greater than 2 (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.188669DS1), suggesting cyclic mechanical stress induces gene expression changes associated with proinflammatory states. These pathways included “TNF alpha signaling via NFκB,” “Inflammatory Response,” “Allograft Rejection,” “IL6-JAK-STAT3 Signaling,” “IL2-STAT5 Signaling,” and “Interferon Gamma Response.”
 Figure 2
Figure 2Dynamic loading for 6 hours led to many changes in gene expression. (A) Schematic of experimental design. (B) Sum-of-slices Z-projection of 20× confocal image of HISMCs on aligned scaffolds stained for F-actin (Phalloidin, magenta) with nuclei labeled with Sytox Green. Scale bar: 100 μm. (C) PCA plot (based on the top 500 most variable genes) showing loaded versus unloaded HISMCs differ significantly in gene expression. Orange dots, loaded; blue dots, unloaded. (D) Volcano plot of differentially expressed genes between loaded versus unloaded HISMCs, from DESeq2 analysis. Log2fold change (log2FC) cutoff > 0.48 or < –0.48 (vertical dotted lines). False Discovery Rate (FDR) cutoff = 0.05 (horizontal dotted line). Orange dots indicate differentially expressed genes (FDR < 0.05) with log2FC > 0.48 (1239 genes up in loaded), and blue dots indicate differential expressed genes (FDR < 0.05) with log2FC < –0.48 (1,261 genes up in unloaded).
Consistent with the hypothesis that loading induced a proinflammatory state, STRING classification using KEGG pathways to characterize the 500 most differentially regulated genes (based on adjusted P values) identified 13 NF-κB pathway genes more abundant in loaded than in unloaded HISMCs (Figure 3A). To determine if this reflected increased NF-κB signaling, we used IHC and discovered more nuclear NF-κB in loaded compared with unloaded HISMCs (Figure 3, B and C). Since NF-κB (NFKB1, NFKB2) also promotes a synthetic phenotype in SMCs by repressing myocardin, the master regulator for SMC contractile phenotype (19), we hypothesized that loaded HISMCs might have a more synthetic phenotype than unloaded HISMCs. Consistent with this hypothesis, loaded HISMCs had higher levels of mRNA for many synthetic SMC phenotype genes compared with unloaded HISMCs, including EREG (20), AREG (21), KLF4 (22), PDGFA (23), EPHA2 (24), ETS1, ETS2, ELF1 (25), POU2F2 (26), and THBS1 (27) (Table 1 and Figure 3, D and E). Loaded HISMCs also had less nuclear MKL2 protein compared with unloaded HISMCs (Figures 3, F and G) and less mRNA encoding MKL2 (log2FC = –0.5, adjusted P = 0.0029) (Figure 3H). MKL2 is a myocardin transcription factor family gene (also called Myocardin Related Transcription Factor B [MRTFB]) that promotes the SMC contractile phenotype (28). Furthermore, loaded HISMCs had less mRNA for CARMN (reported as MIR143HG in Supplemental Table 1) (log2FC = –2.06, adjusted P = 9.68 × 10–5), a long noncoding RNA critical for maintaining visceral SMC contractile function (29) (Figure 3I). Collectively, these data show our cyclic stretching paradigm promotes a synthetic, proinflammatory state in HISMCs, instead of a contractile phenotype.
 Figure 3
Figure 3Loaded HISMCs have increased synthetic gene expression and activation of NF-κB signaling. (A) STRING diagram of KEGG NF-κB pathway analysis. Input included mRNA more abundant in loaded HISMCs with log2 fold change (log2FC) > 0.48 based on bulk RNA-Seq. (B) Representative sum-of-slices Z-projections of confocal images (63× oil objective) showing NF-κB antibody staining (magenta) and Sytox green nuclear staining. Top: Unloaded HISMCs grown on aligned scaffold. Bottom: Loaded HISMCs grown on aligned scaffold. Scale bar: 20 μm. (C) Quantitative analysis of antibody staining demonstrated increased NF-κB nuclear to cytoplasmic ratio in loaded compared with unloaded HISMCs (median [interquartile range] unloaded 1.152 [0.4651], loaded 1.336 [0.8271] (P = 0.0081, Mann Whitney), n = 79 cells for both groups. (D and E) qPCR analyses for mRNA levels of smooth muscle synthetic genes (Amphiregulin, Epiregulin) showed increased Amphiregulin and Epiregulin expression in loaded HISMCs. Amphiregulin (median [interquartile range], unloaded: 0.01114 [0.006101], loaded: 0.4518 [0.1364], P = 0.0286, n = 8). Epiregulin (median [interquartile range], unloaded: 0.0002173 [0.000219], loaded: 0.005386 [0.01303], P = 0.0286, n = 8). (F) Representative sum-of-slices Z-projections of confocal images (63× oil objective) of MKL2 stained HISMCs (red) and Sytox green nuclear staining. Top: Unloaded HISMCs grown on aligned scaffold. Bottom: Loaded HISMCs grown on aligned scaffold. Scale bar: 20 µm. (G) Quantitative analysis of antibody staining demonstrated an increased MKL2 nuclear to cytoplasmic ratio in unloaded compared with loaded HISMCs (median [interquartile range] unloaded 2.435 [1.066], loaded 1.820 [0.731], P < 0.0001, Mann Whitney, n = 65 cells for unloaded and n = 70 cells for loaded HISMCs). (H and I) qPCR analyses for mRNA of smooth muscle contractile genes showed decreased CARMN and MKL2 expression in loaded HISMCs. CARMN (median [interquartile range], unloaded: 0.001717 [0.003256], loaded: 0.0003141 [0.0003979], P = 0.0286, n = 8). MKL2 (median [interquartile range], unloaded: 0.001717 [0.003256], loaded: 0.0003141 [0.0003979], P = 0.0286, n = 8). *P < 0.05, **P < 0.01.
 Table 1
Table 1Smooth muscle synthetic genes differentially expressed in loaded versus unloaded HISMC based on bulk RNA-Seq
These findings are reinforced by STRING classification of the top 500 genes (by adjusted P value) with absolute value log2FC > 0.48 using Gene Ontology (GO) Biological Process pathways (30, 31). In STRING analysis, 30 of these top 500 genes were involved in cytokine signaling (cytokine-mediated signaling pathway, GO:0019221) including IL8, CXCL3, IL11, IL1B, CCL20, PTSG2, CXCL2, IL6, LIF, IL24, CXCL1, CXCL5, and CLCF1 (Figure 4A and Table 2). Many of these cytokines may impair intestinal motility (32–34). To validate RNA-Seq, we used quantitative PCR (qPCR) to analyze mRNA abundance for IL6 (Figure 4B), a major proinflammatory cytokine (35), and IL11 (Figure 4C), which promotes a synthetic phenotype in vascular smooth muscle (34). qPCR showed that IL6 mRNA was 12-fold more abundant (P < 0.001, n = 4) in loaded versus unloaded HISMCs and IL11 mRNA was 55-fold (P = 0.004, n = 4) more abundant in loaded HISMCs. This is similar to the 13.4-fold (log2FC = 3.74) elevation in IL-6 and 43.1-fold (log2FC = 5.43) elevation in IL11 based on RNA-Seq (Table 2). In contrast to mRNA, IL-11 IHC revealed lower protein levels in loaded than in unloaded HISMCs (Figures 4, D and E). In addition, phospho-STAT3, a key IL-6 signaling protein, was not detected in either loaded or unloaded HISMCs by antibody staining (Figure 4F) although we readily detected phospho-STAT3 in human THP-1 macrophages (Supplemental Figure 2). Collectively, these data show dramatic increases in many proinflammatory signaling molecules at the mRNA level after only 6 hours of pathologic stretching.
 Figure 4
Figure 4Supraphysiologic cyclical stretching stimulates production of cytokines, cytokine receptors, and chemokines. (A) STRING diagram of 32 genes (from top 500 differentially expressed genes (Padj < 1 × 10–6, absolute value log2FC > 0.48 out of 1,113 genes meeting this criteria) from bulk RNA-Seq annotated as genes in GO Biological Process cytokine-mediated signaling pathway (GO:0019221) in STRING analysis. (B) qPCR shows 12-fold more IL-6 mRNA in loaded compared with unloaded HISMCs (mean ± SEM unloaded: 0.009601 ± 0,001695, loaded: 0.1195 ± 0.01191, P < 0.0001, n = 4). (C) qPCR shows 55-fold more IL11 mRNA in loaded compared with unloaded HISMCs (mean ± SEM unloaded: 0.02060 ± 0.002797, loaded: 1.136 ± 0.1585, P = 0.0004, n = 4). (D) Quantification of IL-11 IHC shows a small, statistically significant increase in pixel intensity in unloaded compared with loaded HISMCs (median [interquartile range], unloaded: 1,184 [762.6], n = 122; loaded: 1,012 [631.2], n = 99; P = 0.0469, Mann-Whitney). (E) Representative sum-of-slices Z-projections of confocal images of loaded or unloaded HISMCs stained with antibodies to IL11 (63× oil objective, confocal Z-stack). (F) Representative sum-of-slices Z-projections of confocal images of loaded or unloaded HISMCs stained with antibodies to phosphorylated STAT3 (PSTAT3) (63× oil objective, confocal Z-stack). PSTAT3 was not detected in HISMCs under either condition, but PSTAT3 was readily detectable in human monocyte THP-1 cell line (Supplemental Figure 2). *P < 0.05, ***P < 0.001, ****P < 0.0001.
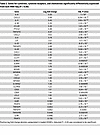 Table 2
Table 2Genes for cytokines, cytokine receptors, and chemokines significantly differentially expressed from bulk RNA-Seq (P < 0.05)
TGF-β superfamily genes are differentially expressed in loaded versus unloaded HISMC. TGF-β signaling has roles in smooth muscle embryogenesis and phenotypic class switching (36). Many TGF-β superfamily genes were differentially regulated by 6 hours of cyclic HISMC loading based on RNA-Seq. Loaded HISMCs had more INHBB, TGFBR1, TGFBR3, SMAD7, TGFB1, BMP2, GREM1, and SMAD1 mRNA and less TMEM100, SMAD6, BAMBI, BMP4, SMAD6, and BAMBI mRNA, compared with unloaded HISMCs (Table 3). qPCR confirmed higher levels of BMP2 (Figure 5A) and GREM1 (Figure 5B) in loaded HISMCs and reduced BMP4 mRNA (Figure 5C) compared with unloaded cells. Since TGF-β and BMP can alter SMC phenotype, we evaluated nuclear to cytoplasmic ratios of signaling proteins that localize to the nucleus after BMP (phospho-SMAD1/5/8) or TGF-β (phospho-SMAD2/3) receptor activation (Figure 5D). Quantitative analysis of IHC showed equivalent nuclear to cytoplasmic ratios of phospho-SMAD2/3 and phospho-SMAD1/5/8 in loaded and unloaded HISMCs (Figure 5, E and F). Collectively, these data indicate that cyclic stretching rapidly alters mRNA levels for many TGF-β superfamily genes, but the signaling pathways that these genes could activate or inhibit were not altered in HISMCs, at least at this early time point.
 Figure 5
Figure 5TGF-β superfamily genes are differentially expressed after HISMC loading. (A) qPCR confirms 5.36× increased BMP2 in loaded HISMCs (mean ± SEM unloaded: 0.02222 ± 0.004256, loaded: 0.1191 ± 0.01708, P = 0.0015, n = 4). (B) qPCR showed GREM1 mRNA is 3.16× increased in loaded HISMCs (mean ± SEM unloaded: 0.0007025 ± 0.0001186, loaded: 0.00289 ± 0.0004599, P = 0.0156, n = 4). (C) BMP4 reverse transcription PCR results confirming 8.63× increased BMP4 expression in unloaded HISMCs (mean ± SEM unloaded: 0.2358 ± 0.07209, loaded: 0.02733 ± 0.008379, P = 0.0284, n = 4). (D) Representative images taken with confocal microscope 63× oil objective of pSMAD 1/5/8 (left) and pSMAD 2/3 (right) in unloaded and loaded HISMCs. (E) IHC quantification of PSMAD 1/5/8 showed no differences in nuclear to cytoplasmic staining between loaded and unloaded HISMCs (median [interquartile range], unloaded: 4.549 [2.937], n = 150; loaded: 4.391 [2.584], n = 88; P = 0.5339, Mann-Whitney). (F) IHC quantification of PSMAD 2/3 showed no differences in nuclear to cytoplasmic ratio between loaded and unloaded HISMCs (median [interquartile range], unloaded: 1.547 [0.471], n = 150; loaded: 1.598 [0.344], n = 88; P = 0.5099, Mann-Whitney).*P < 0.05, **P < 0.01.
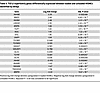 Table 3
Table 3TGF-β superfamily genes differentially expressed between loaded and unloaded HISMCs identified by DESq2
Pathologic loading induces differential expression of guidance molecules and of genes needed for cell-cell and cell-ECM interactions. Cyclic HISMC loading rapidly altered mRNA levels for many ephrins, semaphorins, netrins, and slits (Table 4). In addition to central roles in neurobiology, these axon guidance molecules play key roles in vascular SMC migration, cell proliferation, and inflammation in cardiovascular disease (37). Several mRNAs involved in cell-ECM or cell-cell interactions, with possible roles in mechanosensation, were differentially expressed between loaded and unloaded HISMCs. These included integrins, cadherins, catenins and catenin antagonists, claudins, a tight junction protein, talins, syndecans, an actinin, an adherens junction protein, cell adhesion molecules, and focal adhesion genes (Table 5). Finally, there were differential changes in mRNA encoding many cytoskeletal proteins (Table 6). These changes in gene expression suggest that, in response to cyclic stretching, HISMCs alter cell-cell and cell-ECM interactions, possibly consistent with a transition away from contractile SMC phenotypes.
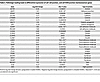 Table 5
Table 5Pathologic loading leads to differential expression of cell-cell junction, and cell-ECM junction mechanosensor genes
Loading induced expression of ligands that could signal to nearby cells. Remarkably, many genes differentially regulated in HISMCs by cyclic loading encode secreted or cell surface ligands that could affect biology of nearby cells by binding cell surface receptors. To identify possible cellular targets for differentially expressed HISMC ligands, we used NicheNet (38) and human bowel single nucleus RNA-Seq data from Drokhlyansky et al. (39). The analysis strategy is summarized in Supplemental Figure 3. NicheNet evaluates potential ligand-receptor interactions, ranking interactions based on ligand-target regulatory potential (incorporating intracellular signaling into regulatory potential scoring). These potential ligand-receptor interactions are represented in Sankey plots (Figures 6 and 7). On the left of each Sankey plot is a ligand whose mRNA is more abundant in loaded HISMCs than in unloaded HISMCs (Figure 6), or conversely, a ligand more abundant in unloaded HISMCs than in loaded HISMCs (Figure 7), based on our data. In the middle column are receptors (from Drokhlyansky et al. data) (39) for ligands differentially expressed in our dataset. On the right of each Sankey plot are genes whose activity or expression is regulated by receptor signaling, according to the NicheNet model. The color of each line indicates the receptor-bearing cell type, based on Drokhlyansky et al. data (39). Figures 6 and 7 show the top 10 prioritized ligands from HISMCs (based on log2FC) for each cell type in a reannotated subset of Drokhlyansky et al. data. Additional data in Supplemental Figure 4 shows genes more abundant in loaded HISMCs for the next 10 prioritized ligands (Figure 6, Figure 7, and Supplemental Figure 4 show more than 10 ligands because top prioritized ligands differ between cell types). For example, BMP4 mRNA is increased in unloaded compared with loaded HISMCs (as we confirmed in Figure 5C). The Sankey plot (Figure 7) shows that receptors for BMP4 (i.e., BMPR1A, BMPR1B, and BMPR2) are expressed in visceral smooth muscle (VisceralSMC_1). However, BMPR1A is also expressed in enteric neurons, macrophage, Fibroblast_1, and epithelial cells, while BMPR1B is expressed in neurons, Interstitial cells of Cajal (ICC), and Fibroblast_1. The coreceptor BMPR2 is expressed in VisceralSMC_1, vascular endothelial cells, enteric neurons, ICC, Fibroblast_1, and epithelial cells, but was not detected in macrophages in Drokhlyansky’s dataset (39). While some differentially expressed HISMC ligands could signal to many adjacent cell types (e.g., FGF18, IL6, IL11, AREG, EREG, BMP2, HBEGF in loaded HISMCs; GDF5, BMP4, EFNA1, EFNA3, EFNA4 in unloaded HISMCs), other differentially expressed HISMC ligands were predicted to signal to only to neurons (TNFSF15, IL16, INHBB in loaded HISMCs; ADM, APLN in unloaded HISMCs) or to neurons, vascular endothelial cells, and macrophages (CSF3 in loaded HISMCs). Notably, differentially expressed HISMC ligands from loaded cells have the largest number of targets in neurons, leading to the intriguing hypothesis that neurons may play an active role in how bowel responds to pathologic mechanical stress. For differentially expressed HISMC ligands in unloaded cells, there were fewer targets identified across all examined bowel cell types compared with the number of targets identified across bowel cell types for differentially expressed HISMC ligands in the loaded cells. Nevertheless, neuronal targets again feature prominently. Note that some possible interactions indicated in Sankey plots may not be biologically relevant (e.g., smooth muscle ICAM might never contact bowel epithelial cells). Nonetheless, these NicheNet analyses suggest that altered mechanical stress induces broad changes in HISMC gene expression and that many differentially expressed genes are likely to bind to receptors, and influence function, of other bowel wall cell types.
 Figure 6
Figure 6NicheNet ligand-receptor analysis using ligands more abundant in loaded HISMCs. Sankey plot showing potential ligand-receptor-target links based on NicheNet’s inferred signaling paths from “top 10” ligands upregulated in loaded HISMC Drokhlyansky et al. (39) receiver cell targets. NicheNet prioritized ligand analysis between secreted ligands more abundant in loaded compared with unloaded HISMCs (left column) and receptors (middle column) and target genes (right column) in reannotated Drokhlyansky et al. receiver cell types was used to infer signaling paths from each ligand to target. Potential ligand-receptor-target links were determined based on inferred signaling paths from NicheNet. See Methods and Supplemental Figure 3 for additional details on NicheNet analysis and process for inferring ligand-receptor-targets paths.
 Figure 7
Figure 7NicheNet ligand-receptor analysis using ligands more abundant in unloaded HISMCs. Sankey plot showing potential ligand-receptor-target links based on NicheNet’s inferred signaling paths from “top 10” ligands upregulated in unloaded HISMC to Drokhlyansky et al. (39) receiver cell targets. NicheNet prioritized ligand analysis between secreted ligands more abundant in unloaded compared with loaded HISMCs (left column) and receptors (middle column) and target genes (right column) in reannotated Drokhlyansky et al. receiver cell types was used to infer signaling paths from each ligand to target. Potential ligand-receptor-target links were determined based on inferred signaling paths from NicheNet. See Methods and Supplemental Figure 3 for additional details on NicheNet analysis and process for inferring ligand-receptor-targets paths.















