Inflammation
Abstract
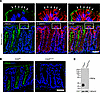
Mammalian skin wounds typically heal with a scar, characterized by fibrotic tissue that disrupts original tissue architecture and function. Therapies that limit fibrosis and promote regenerative healing remain a major unmet clinical need. Rosemary extract, particularly in the form of topical oils and creams, has gained widespread public attention for its purported wound-healing properties. However, its efficacy and mechanism of action remain poorly understood. We show in adult wound healing mouse models that an ethanol-based rosemary extract accelerates the speed of wound healing and mitigates fibrosis. Mechanistically, we identify that carnosic acid, a major bioactive component of rosemary leaves, activates the TRPA1 nociceptor on cutaneous sensory neurons to enhance tissue regeneration. Mice lacking TRPA1 in sensory neurons do not exhibit these pro-regenerative responses, confirming its role as a critical mediator. Together, these findings suggest that topical rosemary extract may represent an effective and accessible therapeutic approach to improve skin repair outcomes.
Authors
Emmanuel Rapp, Jiayi Pang, Borna Saeednia, Stephen Marsh Prouty, Christopher A. Reilly, Thomas H. Leung
Abstract
The gastrointestinal epithelium depends on the apical junctional complex (AJC), composed of tight and adherens junctions, to regulate barrier function. Here, we identify the apical polarity protein Crumbs homolog 3 (CRB3) as an important regulator of AJC assembly and barrier function in intestinal epithelium. Using primary murine colonic epithelial cells (colonoids) from inducible, conditional Crb3-knockout (Crb3ERΔIEC) and control (Crb3fl/fl) mice, we show that CRB3 deficiency compromised barrier function that was associated with a hypercontractile perijunctional actomyosin network and impaired AJC assembly. Loss of CRB3 exacerbated proinflammatory cytokine–induced AJC remodeling, leading to increased intestinal permeability. Crb3ERΔIEC cells exhibited increased RhoA activity and junctional tension, which could be reversed by ROCK-II or myosin II inhibition, restoring junctional architecture. Mechanistically, CRB3A interacts with the actin cytoskeletal linker protein, Merlin (NF2) via its FERM-binding domain, and NF2 knockdown phenocopied CRB3 loss, suggesting their cooperative role in AJC assembly. These findings establish CRB3 and NF2 signaling as key regulators of perijunctional actomyosin contractility and AJC organization during both de novo junctional assembly and inflammation-induced remodeling. This work defines a CRB3- and NF2-dependent pathway by which epithelial cells regulate mechanical tension to coordinate barrier assembly during homeostasis and junctional remodeling under inflammatory stress.
Authors
Shuling Fan, Saranyaraajan Varadarajan, Vicky Garcia-Hernandez, Sven Flemming, Arturo Raya-Sandino, Ben Margolis, Charles A. Parkos, Asma Nusrat
Abstract
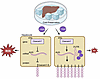
IgA protects the body from invaders in the mucosal sites, but its role in allergic diseases, such as hay fever, is poorly understood. We demonstrate an increased susceptibility to cedar pollen-induced hay fever associated with increasing pollen penetration into the body in IgA-deficient mice, indicating that IgA prevents pollen invasion on the mucosa. We identified Bryostatin 1, an anti-carcinogenic Protein kinase C (PKC) δ activator, as an IgA/IgE class-switching regulator in B cells. Bryostatin 1 enhanced IgA production through induction of germline transcript (GLT) α via PKCδ-MEK/ERK-RUNX1 pathway and suppressed IgE by reducing GLTε through the PKCδ-STAT5-ID2 pathway. Production of Th2 cytokines and eosinophils infiltration in the lungs was also reduced. Furthermore, hay fever alleviation by Bryostatin 1 demonstrated diminished symptoms in mice in vivo three months subsequent to nasal administration.
Authors
Naoki Morita, Kohta Yamamoto, Ryutaro Tamano, Peng Gao, Takahiro Nagatake, Takenori Inomata, Tianxiang Huang, Yasuhiro Yamada, Takahiro Adachi, Manabu Sugai, Keiichi I. Nakayama, Hirotatsu Kojima, Reiko Shinkura
Abstract
SLC26A9 is an epithelial chloride channel that was identified as a genetic modifier of disease severity of cystic fibrosis (CF) and other chronic muco-obstructive lung diseases. However, data on the in vivo role of SLC26A9 function in lung health and disease remain limited. Here, we investigated the effect of genetic deletion of Slc26a9 (Slc26a9-/-) on the pulmonary phenotype of neonatal mice. We found that lack of Slc26a9 causes severe neonatal respiratory distress with high mortality. Histology, immunohistochemistry and micro-computed tomography imaging studies identified airway obstruction with MUC5B-positive mucus plugs in neonatal Slc26a9-/- mice. Bioelectric measurements demonstrated a reduced transepithelial potential difference indicative of reduced chloride secretion across tracheal explants of neonatal Slc26a9-/- compared to wild-type mice. In addition, neonatal Slc26a9-/- mice displayed hypoxic degeneration of airway epithelial cells associated with sterile neutrophilic airway inflammation. Collectively, our data show that SLC26A9-mediated chloride secretion is critical for proper mucociliary clearance, respiratory function and survival after birth, and identify a novel role of SLC26A9 in neonatal adaptation during the transition from fetal to neonatal life.
Authors
Pamela Millar-Büchner, Johanna J. Salomon, Julia Duerr, Stephan Spahn, Pinelopi Anagnostopoulou, Willi L. Wagner, Mark O. Wielpuetz, Hermann-Josef Gröne, Anita Balázs, Marcus A. Mall
Abstract
Insulin resistance impairs benefits of lipid-lowering treatment as evidenced by higher cardiovascular risk in individuals with type 2 diabetes versus those without. Because platelet activity is higher in insulin-resistant patients and promotes atherosclerosis progression, we questioned whether platelets impair inflammation resolution in plaques during lipid-lowering. In mice with obesity and insulin resistance, we induced advanced plaques, then implemented lipid-lowering to promote atherosclerotic plaque inflammation-resolution. Concurrently, mice were treated with either platelet-depleting or control antibodies for 3 weeks. Platelet activation and insulin resistance were unaffected by lipid-lowering. Both antibody-treated groups showed reduced plaque macrophages, but plaque cellular and structural composition differed. In platelet-depleted mice, scRNA seq revealed dampened inflammatory gene expression in plaque macrophages and an expansion of a subset of Fcgr4+ macrophages having features of inflammation-resolving, phagocytic cells. Necrotic core size was smaller and collagen content greater, resembling stable human plaques. Consistent with the mouse results, clinical data showed that patients with lower platelet counts had decreased pro-inflammatory signaling pathways in circulating non-classical monocytes after lipid-lowering. These findings highlight that platelets hinder inflammation-resolution in atherosclerosis during lipid-lowering treatment. Identifying novel platelet-targeted therapies following lipid-lowering treatment in individuals with insulin resistance may be a promising therapeutic approach to promote atherosclerotic plaque inflammation-resolution.
Authors
Maria Laskou, Sofie Delbare, Michael Gildea, Ada Weinstock, Vitor De Moura Virginio, Maxwell La Forest, Franziska Krautter, Casey Donahoe, Letizia Amadori, Natalia Eberhardt, Tessa J. Barrett, Chiara Giannarelli, Jeffrey S. Berger, Edward A. Fisher
Abstract
Coronary artery disease (CAD) is the leading cause of mortality worldwide, with macrophages playing a central role in shaping the inflammatory environment through cytokines, chemokines, and other mediators. Long noncoding RNAs (lncRNAs) are emerging as key regulators of cellular processes due to their interactions with DNA, RNA, microRNAs, and proteins, positioning them as promising therapeutic targets. Through integrative transcriptomic analysis, we identified SPANXA2-OT1 as a primate-specific lncRNA with a potential role in macrophage-mediated inflammation in CAD. Functional studies in primary human macrophages demonstrated that SPANXA2-OT1 is induced by inflammatory stimulation, localized to the cytoplasm, and exerts regulatory effects on chemokine expression and macrophage chemotaxis. Mechanistically, SPANXA2-OT1 acts as a molecular sponge for microRNA-338, thereby influencing the expression of interleukin-8 (IL-8), a critical mediator of monocyte recruitment and inflammatory signaling. Collectively, these findings establish SPANXA2-OT1 as a human-specific regulator of inflammatory pathways in CAD and highlight its translational potential as both a biomarker and therapeutic target.
Authors
Prabhash Kumar Jha, Sarvesh Chelvanambi, Yuto Nakamura, Lucas Yuji Umesaki Itto, Aatira Vijay, Adrien Lupieri, Miguel Cantadori Barbeiro, Thanh-Dat Le, Caio Borges Nascimento, Taku Kasai, Mary C. Whelan, Daiki Hosokawa, Dakota Becker-Greene, Sasha A. Singh, Elena Aikawa, Shizuka Uchida, Masanori Aikawa

Abstract
Clinical trials have identified 2 distinct eosinophilic esophagitis (EoE) treatment phenotypes: those that show proton pump inhibitor (PPI) responsiveness (PPI-R) and those that show PPI unresponsiveness (PPI-UR). Comprehensive clinical, endoscopic, and RNA-Seq analyses of patients with EoE prior to and following PPI therapy have not previously been performed to our knowledge. We showed that clinical, endoscopic, and histologic evaluation of esophageal biopsies from pediatric PPI-R and PPI-UR individuals with EoE prior to PPI therapy (diagnosis) were indistinguishable. RNA-Seq analyses revealed common immune and inflammatory transcriptional signatures in both PPI-R EoE and PPI-UR EoE esophageal biopsy samples at diagnosis and distinct signatures enriched for processes related to neuropeptide signaling and cell cycle and division. PPI therapy induced histologic, endoscopic, and transcriptional remission in PPI-R EoE, but not in PPI-UR EoE. Persistent disease in PPI-UR EoE was associated with the presence of Th2 inflammatory and dedifferentiated esophageal epithelial transcriptomic signatures, while PPI-R EoE revealed genes enriched in cellular responses to LPS, host defense against viruses, and type I IFN signaling. In silico analyses identified common and unique EoE disease gene drivers in PPI-R and PPI-UR EoE. These studies indicate that the 2 EoE phenotypes have unique transcriptomic elements that underlie the molecular nature of PPI-R and PPI-UR EoE disease.
Authors
Somdutta Chakraborty, Ankit Sharma, Sahiti Marella, Christian F. Rizza, Patrick A. O’Brien, Varsha Ganesan, Gila Idelman, Susie Min, Mayee Chen, Talaya McCright-Gill, Nancy Gonzalez, Alexandros D. Polydorides, Paul S. Foster, Simon P. Hogan, Mirna Chehade
Abstract
Glomerular inflammation and podocyte loss are the hallmarks of chronic kidney disease (CKD) progression. Understanding how podocytes and their microenvironment regulate inflammation is critical for developing effective therapies. In this study, we identified C-C chemokine ligand 5 (CCL5) as an inflammatory mediator elevated in injured podocytes, based on analyses of both human kidney biopsies and mouse models of CKD. We discovered that CCL5 exerts paradoxical effects in nephropathy: while it protects podocytes in vitro, it exacerbates glomerular injury in vivo. Recombinant CCL5 and podocyte-specific CCL5 overexpression promoted cell survival and reduced apoptosis in cultured podocytes. However, in Adriamycin-induced nephropathy, CCL5 worsened glomerular injury, increasing proteinuria, glomerulosclerosis, and podocyte loss. Bone marrow (BM) transplantation experiments revealed that CCL5 in BM-derived cells—not kidney-resident cells—drove disease progression. CCL5 deficiency in BM-derived cells conferred protection by increasing reparative M2 macrophages, whereas endogenous CCL5 promoted M1 polarization, inhibited M2 differentiation, and triggered M2-to-M1 transition. These findings demonstrate that while CCL5 supports podocyte survival, its expression in BM-derived cells promotes inflammatory macrophage phenotypes and glomerular injury. The harmful immune effects of CCL5 in BM-derived cells outweigh its podocyte-protective role, highlighting the importance of cell-targeted strategies to mitigate kidney damage.
Authors
Ika N. Kadariswantiningsih, Issei Okunaga, Kaho Yamasaki, Maulana A. Empitu, Hiroyuki Yamada, Shin-ichi Makino, Akitsu Hotta, Hideo Yagita, Masashi Aizawa, Ryo Koyama-Nasu, Motoko Y. Kimura, Narihito Tatsumoto, Katsuhiko Asanuma
Abstract
Hepatic ischemia-reperfusion injury (IRI) disrupts cellular signaling pathways and contributes to early allograft dysfunction (EAD) in orthotopic liver transplantation (OLT). In this study, we found that the hepatic RNA binding protein Human Antigen R (HuR) regulated the 3′ untranslated region (UTR) of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 (Ceacam1) following ischemic stress. Hepatocyte-specific preinjury HuR-null mice exhibited elevated LDH-5 isoenzyme activity and reduced Ceacam1-S expression, reflecting tissue-specific injury. In situ hybridization demonstrated that the stability of Ceacam1 mRNA depended on HuR. Luciferase assays identified Ceacam1 3′UTR cis-elements responsive to high oxygen tension. HuR-targeting short-activating RNAs (saRNAs) preferentially induced the alternative splicing of Ceacam1-S. Antisense oligos directed to the Ceacam1 3′UTR protected WT mice against acute liver injury. In the clinical arm, increased HuR and CEACAM1 expression were associated with reduced proinflammatory phenotype and a lower incidence of EAD in patients with OLT (n = 164). Human discarded livers with elevated ELAVL1/CEACAM1 levels correlated with improved tissue homeostasis. These findings suggest that HuR regulation of Ceacam1 represents a key determinant of donor tissue quality and offers a potential target for future therapeutic strategies in OLT recipients.
Authors
Brian Cheng, Tristan D. Tibbe, Siyuan Yao, Megan Wei, Zeriel Y. Wong, Taylor Torgerson, Richard Chiu, Aanchal S. Kasargod, Kojiro Nakamura, Monica Cappelletti, Myung Sim, Douglas G. Farmer, Fady Kaldas, Jerzy W. Kupiec-Weglinski, Kenneth J. Dery
Abstract
The intestinal mucosal epithelium forms a barrier between luminal contents and the body. microRNAs (miRNAs) regulate mucosal homeostasis by controlling inflammatory responses and structural integrity. Here, we discovered a protective role for miR147 in intestinal inflammation using a miR147tdTomato reporter mouse. miR147 was enriched in the intestines, with the highest expression in the colonic epithelial cells at the luminal surface, with prominent expression in differentiated enterocytes. Mice with general or intestinal epithelial deletion of miR147 showed increased intestinal inflammation and diminished mucosal healing during colitis. RNA sequencing of miR147-deficient cells showed dysregulated immune signaling, with upregulated pro-inflammatory cytokine pathways and reduced type I interferon responses and revealed Ndufa4 as a likely miR147 target. Ndufa4, a mitochondrial protein regulating energy metabolism and inflammation, is elevated at the crypt base, inversely correlating with miR147. Mice lacking the miR147 binding site in Ndufa4’s 3′ untranslated region phenocopied miR147-deficient mice during colitis. Spatial and single-cell transcriptomic analyses in murine and human colons showed mutually exclusive miR147 and Ndufa4 expression, consistent with a regulatory relationship in epithelial differentiation and metabolism. These findings underscore miR147’s role in intestinal homeostasis and mucosal healing, suggesting it as a therapeutic target for inflammatory bowel disease.
Authors
Agnieszka K. Czopik, Arash Dabiri, Chia-Hao Tung, Victoria Vaughn, Xiangsheng Huang, Jinlian Wang, Hui Li, Nicolas F. Moreno, Natalia V. Piwko, Katherine Figarella, Hongfang Liu, Zhongming Zhao, Xiaoyi Yuan, Holger K. Eltzschig
No posts were found with this tag.


