Research ArticleInflammationPulmonology
Open Access | ![]() 10.1172/jci.insight.196018
10.1172/jci.insight.196018
Transcriptomic and functional responses of the cystic fibrosis airway epithelium to CFTR modulator therapy
Eszter K. Vladar,1 Austin E. Gillen,1,2 Sangya Yadav,1 Mikayla R. Murphree,1 David Baraghoshi,3 J. Kirk Harris,4 Elmar Pruesse,1,5,6 Sierra S. Niemiec,7 Alexandra W.M. Wilson,8 Katherine B. Hisert,9 Stephen M. Humphries,10 Matthew Strand,3 David A. Lynch,10 Max A. Seibold,1,5,6 Daniel M. Beswick,11 and Jennifer L. Taylor-Cousar6,9
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by
Vladar, E.
in:
PubMed
|
Google Scholar
|

1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Gillen, A. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Yadav, S. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Murphree, M. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Baraghoshi, D. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Harris, J. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Pruesse, E. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Niemiec, S. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Wilson, A. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Hisert, K. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Humphries, S. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Strand, M. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Lynch, D. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by
Seibold, M.
in:
PubMed
|
Google Scholar
|
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by Beswick, D. in: PubMed | Google Scholar
1Department of Medicine, University of Colorado, Aurora, Colorado, USA.
2Rocky Mountain Regional Veterans Affairs Medical Center, Aurora, Colorado, USA.
3Division of Biostatistics, National Jewish Health, Denver, Colorado, USA.
4Departments of Pediatrics, University of Colorado, Aurora, Colorado, USA.
5Center for Genes, Environment, and Health, and
6Department of Pediatrics, National Jewish Health, Denver, Colorado, USA.
7Department of Biostatistics & Informatics, University of Colorado, Aurora, Colorado, USA.
8Clinical Research Services,
9Department of Medicine, and
10Department of Radiology, National Jewish Health, Denver, Colorado, USA.
11Department of Otolaryngology-Head and Neck Surgery, UCLA, Los Angeles, California, USA.
Address correspondence to: Eszter K. Vladar, University of Colorado School of Medicine, 12700 E. 19th Avenue, MS C272, Aurora, Colorado 80045, USA. Email: eszter.vladar@cuanschutz.edu.
Authorship note: DMB and JLTC have been designated as co–senior authors.
Find articles by
Taylor-Cousar, J.
in:
PubMed
|
Google Scholar
|
Published November 10, 2025 - More info
JCI Insight. 2025;10(21):e196018. https://doi.org/10.1172/jci.insight.196018.
© 2025 Vladar et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: June 25, 2025; Accepted: September 23, 2025
-
Results
Extensive longitudinal transcriptional changes underlie the sinonasal mucosal response to initiating ETI in an adult cohort of pwCF. To delineate longitudinal changes in cellular programming and function within the sinonasal airway epithelium in response to ETI in a cohort of adult pwCF with established lung and sinus disease (16–23), participants were recruited for nasal brushing of the inferior turbinates at baseline and at 6 mo. and 2 yr. after initiating ETI therapy. Samples were obtained from a total of 30 participants, including 28, 24, and 23 participants at baseline, 6 mo., and 2 yr., respectively (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.196018DS1), representing 21 complete time courses, 24 baseline–6 mo. pairs, 22 baseline–2 yr. pairs, and 21 6 mo.–2 yr. pairs. Cells obtained from the left nostril were banked for subsequent primary airway epithelial cell culture, while cells obtained from the right nostril were subjected to whole transcriptome RNA sequencing (Figure 1A). Principal component analysis (PCA) of the transcriptomic data identified two 2 yr. samples as potential outliers (Supplemental Figure 1); therefore, analyses were carried out both with and without these samples. Differential gene expression testing (adj. P < 0.05) across the experimental time points yielded a total of 919 genes including the outliers and 806 excluding the outliers (Supplemental Table 2). Including or excluding outliers, the most highly enriched molecular pathways were related to neutrophilic inflammation (SERPINA1, IL1A, IL1RN, TLR4, PECAM1, NLRP3, FCGR2A), cell cycle regulation (AURKB, CENPF, FOXM1), and cell signaling (EREG, LRP5, NUMB) (Figure 1B). Genes related to motile cilium biogenesis (CEP55, CETN3, DNAH14) and squamous/cornified epithelium (IVL, SPRR3, KRT13) were also significantly differentially expressed. Including outliers yielded additional immune function–related genes, including those responsive to viral infection (CCL20, CXCR5, MX2), suggesting that these 2 samples may represent a more highly inflammatory state. Of note, one of the participants reported flu-like symptoms at the time of sampling. Downstream analyses exclude outliers unless otherwise noted. One additional participant underwent lung transplantation between the 6 mo. and 2 yr. time points, so the 2 yr. time point for this participant was excluded from pulmonary function–related comparisons (Supplemental Table 1). In sum, time course gene expression signatures in this cohort capture a drastically changing molecular landscape related to inflammatory epithelial remodeling in the CF sinonasal airways in response to ETI.
 Figure 1
Figure 1Sinonasal differential gene expression in response to ETI therapy. (A) Study schematic with time points. (B) Pathway enrichments for genes differentially expressed across the entire time course. (C) Pathway enrichments for up- and downregulated genes from baseline to 6 mo. (D) Plot of Pearson’s correlation of gene expression related to cilia and ciliated cells and airway epithelial inflammation. (E) Pathway enrichments for up- and downregulated genes from 6 mo. to 2 yr.
Sinonasal transcriptional responses from baseline to 6 mo. reflect decreased neutrophilic inflammation and improved epithelial remodeling. We hypothesized that the rapid and marked improvement in CF airway disease upon initiating ETI is accompanied by changes in gene expression that reflect decreased inflammation and normalization of epithelial structure and function. Consistent with this hypothesis, we observed 865 downregulated genes (adj. P < 0.05) from baseline to 6 mo. that were dominated by those related to neutrophil degranulation, neutrophil extracellular trap formation, cytokine production and signaling, and chemotaxis, and included critical drivers of CF airway inflammation such as CXCL8, CXCR1, CXCR2, TLR4, PTGS2, and IL1R2 (2). Genes related to cell senescence and senescence-associated secretory phenotype (24) (SASP; FOS, CDKN2D, MAPK3) were also markedly downregulated (Figure 1C and Supplemental Table 3). The 631 baseline to 6 mo. upregulated genes (adj. P < 0.05) were involved in various metabolic processes (GLS2, DHDH, ACADL), organelle biogenesis and function, including motile cilium biogenesis (WDPCP, CEP57, BBS10), and cell cycle control (CDK4, ORC3, ANAPC16) (Figure 1C and Supplemental Table 3). These data identify molecular factors underlying the widely reported attenuation of inflammatory drivers of disease after ETI therapy, and further indicate a shift toward normalization of airway epithelial structure and function. This finding was further supported by a significant negative correlation (r = –0.53, P = 0.0071) in expression between genes related to motile cilia and ciliated cells (25) and genes related to sinonasal airway epithelial inflammation (26) for the baseline to 6 mo. span (Figure 1D).
To delineate global cellular and molecular processes involved in the transcriptional response to ETI, hierarchical clustering based on shared patterns of expression was used to identify 7 gene clusters (Figure 2A and Supplemental Table 4). The 3 clusters related to inflammation exhibited strongly decreased expression from baseline to 6 mo. These clusters included genes related to neutrophil degranulation and cytokine signaling (Cluster 2: FCGR2A, FGR, LCP2, CD53, CSF3R), T cell activation (Cluster 5: TNFSF8, IL1A, IL2RG), and inflammatory signaling (Cluster 7: NLRP3, NLRP6, TLR4, TLR8). Three clusters were related epithelial structure and function. Two clusters were upregulated from baseline to 6 mo. and included genes related to motile cilia and developmental signaling (Cluster 1: TUBE1, CFAP47, LRP5, GSK3A, NUMB) and ion homeostasis and transport (Cluster 3: SCN4B, SLC6A13, SLC5A9, BEST4). Conversely, the cluster related to squamous/cornified epithelium and keratinization (Cluster 4: IVL, KRT13, SPRR3) was downregulated from baseline to 6 mo. Finally, a cluster related cell cycle control and proliferation (Cluster 6: TOP2A, CENPI, SPC24, ESCO2) was mostly downregulated from baseline to 6 mo.
 Figure 2
Figure 2Decreased inflammatory gene expression reverses after long-term ETI therapy. (A) Hierarchical clustering of differentially expressed genes during the time course. Heatmap shows the mean-centered log2(fold change) in expression. (B) Box-and-whisker plots of select individual genes of interest related to immune and epithelial cell processes. Significance assessed by 1-way ANOVA with Dunnett’s post hoc test, 2 yr. transcriptional outliers excluded.
To identify functional gene networks based on shared expression throughout the time course, weighted gene coexpression network analysis (WGCNA) (27) was used to identify 12 coexpression modules (Table 1, Supplemental Figure 2, and Supplemental Table 5). The 5 inflammation- and immune function–related modules shared a pattern of decreased expression from baseline to 6 mo. The brown module was strongly enriched in neutrophilic inflammation–related genes (CXCL8, TLR4, S100A8, IL1R2), while the tan module contained genes belonging to the Toll-like receptor (TLR), IL-1, and TGF-β signaling pathways (THBS1, MEF2C, MAPK14, IL6R). The blue, red, and black modules corresponded to functions related to innate immunity (MYD88, LCN2, IRAK2, APOE), humoral adaptive immunity (CD79A, LAMP3, IGHA1, IGKC), and cell-mediated immunity (CD4, CD8A, IL21R, IL32), respectively. Three modules described the epithelial remodeling response with increased expression from baseline to 6 mo. The turquoise and magenta modules corresponded to motile ciliogenesis (FOXJ1, RFX3, IFT88, DNAH5) and ciliary signaling (PKD1, WDR90, AHI1, CCDC57), respectively. The yellow epithelial signaling module contained genes related to the Wnt, Hippo, epidermal growth factor, and other developmental signaling pathways (WNT3A, WNT2B, YAP1, EGFR) key to epithelial differentiation. The remaining modules displayed mixed patterns of expression and corresponded to lipid biosynthesis, lysosomal function, and ion homeostasis (green: SMPD1, FABP5, LAMP1, SCNN1A), mRNA splicing (pink: SFSWAP, SRRM2, PRPF38B, RSRP1), the cytoskeleton (green-yellow: TUBG1, CKAP2, BFSP1, CILK1), and organelle function (purple: RPL5, RPS15, EIF3E, NOP53). In summary, these data indicate that initiating ETI therapy results in wide-ranging cellular programming changes in both the epithelial and immune compartments that reflect substantially decreased inflammation accompanied by improved airway epithelial structure and mucociliary function for the baseline to 6 mo. timespan.
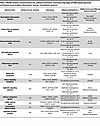 Table 1
Table 1WGCNA networks characterized by size, pathway enrichments, and human lung single-cell RNA sequencing cluster enrichments reveal multiple inflammatory, immune, and epithelial networks
Sinonasal transcriptional response from 6 mo. to 2 yr. reflects increased neutrophilic inflammation. Interestingly, the 6 mo. to 2 yr. gene expression changes revealed 595 upregulated genes (adj. P < 0.05), which indicated a modest, but significant, upregulation of inflammatory pathways, including genes that were downregulated from baseline to 6 mo. (Figure 1E and Supplemental Table 3). This resurgence was also illustrated at the single gene expression level (CXCR4, SERPINA1, TGFB1, TNF, and others; Figure 2B). It was further corroborated by hierarchical gene clustering analysis where Clusters 2, 5, and 7 comprised of genes related to neutrophil degranulation and cytokine signaling, T cell activation, and inflammatory signaling, respectively, increased from 6 mo. to 2 yr. (Figure 2A). Moreover, the red, black, and tan WGCNA modules related to humoral adaptive immunity, cell-mediated immunity, and inflammatory signaling pathways, respectively, also displayed increased expression from 6 mo. to 2 yr. (Table 1). Finally, increased inflammatory gene expression was also observed at the cohort level by PCA, which showed that the displacement along the first principal component observed at 6 mo. compared with baseline was partially reversed at 2 yr., where the first principal component was defined by inflammation- and immune cell–related genes (IL1B, CXCL8, FCGR3B, CXCR1, CXCR2; Supplemental Figure 3, A and B, and Supplemental Table 6).
Interestingly, despite increased inflammatory gene expression, epithelial gene expression, which increased from baseline to 6 mo. to reflect improved epithelial structure and function, did not decrease from 6 mo. to 2 yr. Hierarchical clustering indicated no substantial changes in gene expression in Clusters 1, 6, and 4 related to motile cilia and ciliated cells, cell cycle and proliferation, or squamous epithelium, respectively (Figure 2A and Supplemental Table 4). This was consistent with no changes in the motile ciliogenesis and ciliary signaling (turquoise and magenta) or the epithelial signaling (yellow) WGCNA modules (Table 1, Supplemental Figure 2, and Supplemental Table 5). In sum, our data for the baseline to 6 mo. to 2 yr. timespan indicate that the initial, drastic decrease in inflammatory gene expression upon initiating ETI eventually began to resurge, although this resurgence was not accompanied by gene expression reflecting worsened epithelial structure and function. Of note, baseline sample collection spanned a 10-month period from September to July; thus, gene expression differences at 6 mo. or 2 yr. were unlikely to be driven by seasonal differences in exposures, rather they reflect the ongoing response to ETI.
Changes in cellular composition are likely key drivers of the sinonasal airway response to ETI. To understand what cell types may be driving the transcriptional response to ETI, we mapped the differentially expressed genes across the time course (Supplemental Table 2) to cell types and states identified in a healthy human lung single-cell RNA sequencing dataset (28). Nasal brush biopsies are mainly comprised of epithelial cells but also contain the immune cells that infiltrate or transit over the sampled epithelium. Correspondingly, differentially expressed genes mapped to epithelial cells including basal stem, secretory (mucous and non-mucous), and ciliated cell types and immune cells including neutrophils, mononuclear phagocytes (monocytes, macrophages, dendritic cells [DCs]), T cells, and other immune cell types (B cells, mast cells, eosinophils; Figure 3A). Consistent with the extensive transcriptional changes in immune and inflammatory processes, most (57%) of the mapped transcripts corresponded to immune cells, with the majority identified broadly as mononuclear phagocytes and neutrophils (both at 17%, Figure 3A). Furthermore, 29% of the differentially expressed transcripts across the time course mapped to ciliated cells, reflecting the extensive transcriptomic changes related to motile cilia upon ETI treatment (Figure 2A and Table 1).
 Figure 3
Figure 3Cell type proportionality changes reflect a changing cellular landscape after ETI therapy. (A) Cell type enrichments of genes differentially expressed during the time course suggest major changes in mononuclear phagocytes, neutrophils, and ciliated cells. Computational deconvolution of immune cell type proportions based on healthy human lung (B) and CF sputum–derived (C) single-cell RNA sequencing reference datasets indicates an initial decrease in phagocytes and an increase in T cells upon ETI, which begins to reverse in the 6 mo. to 2 yr. span. Graphs show box-and-whisker plots with a Kruskal-Wallis test (B and C).
Published healthy lung and CF sputum (Figure 3, B and C, and Supplemental Figure 4A) single-cell RNA sequencing reference datasets (28, 29) were used to computationally deconvolve the bulk RNA sequencing data to identify represented cell types and estimate cell type proportion changes over the time course. The 2 reference datasets contain partially overlapping immune cell types and produced similar results. The proportion of neutrophils trended toward decreasing and the proportion of monocyte/macrophage populations decreased significantly from baseline to 6 mo. and both trended toward increasing across the 6 mo. to 2 yr. time point. The opposite change was observed for T cells and plasmacytoid DCs (pDCs), which showed a significant increase from the baseline to 6 mo. and decrease across the 6 mo. to 2 yr. time points. Including the two 2 yr. transcriptional outliers in the analysis revealed that these samples were distinguished by an extremely high predicted proportion of neutrophils, monocytes, and macrophages, and a very low proportion of T cells, DCs, and epithelial cells consistent with the strong inflammatory signature in these samples (Figure 1B and Supplemental Figure 4B).
Epithelial cell changes showed a trend toward an increased proportion of ciliated cells from baseline to 6 mo. and no change between 6 mo. and 2 yr. and secretory cell types trended toward a decrease from baseline to 6 mo. and increase from 6 mo. to 2 yr. (Supplemental Figure 4A). The fraction of basal cells trended toward decreasing from 6 mo. to 2 yr., possibly reflecting an epithelium with reduced injury repair and turnover, which is consistent with the decrease in the proliferative gene signatures over the same timeframe (Figure 2A). Although future experiments are needed to validate the computational inference of cell type proportionality changes, these data suggest that there may be reduced recruitment of phagocytes and increased recruitment of T cells to the sinonasal airways in response to ETI, which begins to reverse despite ongoing therapy.
Sinonasal epithelial transcriptomic responses correlate with airway disease clinical outcomes following ETI treatment. Previously published clinical outcomes for this cohort of pwCF demonstrated substantial improvements in CF-related CRS and in lung function from baseline to 6 mo., with no significant cohort level changes from 6 mo. to 2 yr. (16–23). The percentage of the opacified 3-dimensional sinus volume calculated from computed tomography (%SO) (30) decreased an average of 21.7 points from baseline to 6 mo. (62.5 ± 4.0 to 40.8 ± 3.1, P < 0.0001) and was not significantly different from 6 mo. to 2 yr. (40.8 ± 3.1 to 36.6 ± 3.2, P = 0.82; Supplemental Figure 5A). Association analysis adjusted for time between %SO levels and differentially expressed genes across the time course (Supplemental Table 2) showed that lower %SO, an indication of reduced CRS severity, was significantly (adj. P < 0.05) associated with the decreased expression of 470 genes. These were enriched in pathways related to neutrophil activation (FCGR2A, CXCR1, CXCR2, SERPINA1), neutrophil extracellular trap formation (AQP9, C5AR1, ITGAM, PADI4), and cytokine production and signaling (IL1A, CXCL8, TNF, TGFB1; Figure 4, A and B, and Supplemental Table 7). The top 25 genes whose reduced expression was most strongly associated with lower %SO included JAK1 (Janus kinase 1), STAT6 (signal transducer and activator of transcription 6), and GNS (glucosamine N-acetyl-6-sulfatase), which may be related to downregulation of airway epithelial mucus hypersecretion, a key target in CF disease attenuation by ETI therapy. Lower %SO was also significantly (adj. P < 0.05) associated with increased expression of 100 genes that were enriched in pathways related to RNA processing and protein translation (MRPS25, TSEN34, PCBP1, RPS11) and mitochondrial function and oxidative stress response (TOMM7, LDHD, SUCLG2, NDUFAF2; Figure 4A). Increased expression of these genes may reflect normalization of epithelial structure and function in response to ETI. Association analysis between gene expression and %SO at distinct time points (baseline, 6 mo., or 2 yr.) or the change in %SO between specific time points (baseline to 6 mo. or 2 yr., or 6 mo. to 2 yr.) produced strongly overlapping outcomes with the above at P < 0.05 (Supplemental Figure 5, B and C, and Supplemental Table 7).
 Figure 4
Figure 4Time course gene expression is associated with clinical outcomes related to sinus disease and respiratory function after ETI therapy. (A) Pathway enrichments for genes whose decreased (left) or increased (right) expression is associated with lower %SO. (B) Association plots for select single genes of interest and %SO across the time course. The 6 mo. and 2 yr. data are nearly overlapping for RHOA and MPRS25. Each panel shows for a particular gene the predicted %SO (95% CI) as a function of gene expression (VST) derived from the linear mixed effects models that adjusted for time (excluding the time by gene expression interaction). (C) Pathway enrichments for genes whose decreased (left) or increased (right) expression across the time course is associated with higher ppFEV1.
ppFEV1, the chief metric of lung function in pwCF, rose an average of 11.9 points from baseline to 6 mo. (65.6 ± 4.7 to 77.5 ± 5.7, P = 0.01) and was not significantly different from 6 mo. to 2 yr. (17) (77.5 ± 5.7 to 79.8 ± 5.2, P = 0.36; Supplemental Figure 5A). ppFEV1 association analysis yielded only a few genes that passed the significance threshold of an adjusted P value of less than 0.05. However, due to the modest sample size and the complexity of the ppFEV1 metric, genes with an unadjusted P value of less than 0.05 may still represent biologically meaningful candidates. Reduced expression of 93 genes was associated with higher ppFEV1, an indication of better lung function. These were enriched in pathways related to squamous/cornified epithelium (KRT13, DSC3, DSG3, EVPL) and IL-1 family signaling (IL1RL1, CSF1R, RELA, PRKACA; Figure 4C and Supplemental Table 8). The 100 genes whose increased expression was associated with higher ppFEV1 included those related to motile ciliogenesis (CEP44, IFT20, KIZ, TEKTIP1) and mitochondrial oxidative phosphorylation (SUCLG2, ISCA1, SDHAF4, LYRM2; Figure 4C and Supplemental Table 8). These data suggest that increased lung function may be associated with a compositionally more normal epithelium with more ciliated and fewer squamous/cornified cells and lower IL-1–dependent inflammation.
Baseline sinonasal epithelial gene expression may predict clinical response to long-term ETI therapy. Association between baseline gene expression and 6 mo. or 2 yr. %SO and ppFEV1 was tested with the caveat that about half of the participants (16 of 30, Supplemental Table 1) had received less effective CFTR modulators prior to ETI. A greater baseline to 6 mo. decrease in %SO was significantly associated (P < 0.05) with higher baseline expression of genes related to innate immune response and inflammatory signaling (OSM, PTX3, S100A12, PGLYRP1, GPR84) and actin cytoskeleton and intracellular trafficking (RHOA, VASP, RAB5C, IQGAP1; Figure 5A and Supplemental Table 9). A similar set of genes associated with a greater baseline to 2 yr. decrease in %SO included those related to innate immune response (ORM1, PTX3, PGLYRP1), immune cell recruitment and trafficking (GPR84, CXCR4, ICAM1, PECAM1), cytokine signaling (JAK1, STAT5A, NFKB2, TGFB1, IL1A), oxidative stress and mitochondrial function (SOD2, MICU1, NADK2, GPD2), and actin cytoskeleton (RHOA, CD14, FGF11, NRAS; Figure 5A and Supplemental Table 9). Meanwhile, lower baseline expression of genes related to protein translation (EIF3G, ANKZF1, SAYSD1, MRPS25) was correlated with a greater decrease in %SO from baseline to 6 mo. and protein translation (EEF1D, EIF2D, RPL4, RPL5), and Notch signaling (YBX1, MIB2, SPTAN1, ACIN1, ITGB1BP1) from baseline to 2 yr. (Supplemental Figure 6A and Supplemental Table 9). WGCNA modules (Table 1) also identified a correlation (P < 0.05) between absolute %SO values at 6 mo. or 2 yr. or changes in %SO from baseline to 6 mo. or 2 yr. Significant correlations were identified for the red (humoral adaptive immunity), tan (inflammatory signaling), brown (neutrophilic inflammation), and purple (organelle function) modules (Table 2). In addition to multiple inflammation-related genes, both analyses revealed a correlation between genes in the Notch signaling pathway, a central regulator of airway epithelial cell fate and %SO outcomes (gene based approach: YBX1, RPS27A, MIB2, SPTAN1, ACIN1, ITGB1BP1, EPN2, TSPAN15; WGCNA based approach: NLE1, POGLUT1, OTUD5, PSMD2, TLE4, HDAC1, ATXN1). While additional validation and mechanistic studies are needed, these data suggest that baseline Notch activity may be correlated with CRS outcomes after ETI.
 Figure 5
Figure 5Sinonasal airway epithelial gene expression at baseline is associated with clinical outcomes related to sinus disease and respiratory function after ETI therapy. (A) Pathway enrichments for genes whose higher baseline expression is associated with a larger decrease in %SO from baseline to 6 mo. (left) and 2 yr. (right). (B) Pathway enrichments for genes whose higher baseline expression is associated with a larger increase in ppFEV1 from baseline to 6 mo. (left) and 2 yr. (right).
Association analysis revealed that a greater baseline to 6 mo. increase in ppFEV1 was significantly associated (P < 0.05) with higher baseline expression of genes related to immune response and inflammation (MIF, IL1RL1, CSF1R, RELA, S100A2, S100A10) and actin cytoskeleton regulation (RHOA, CFL1, SPTAN1, CAPG, FLII; Figure 5B and Supplemental Table 10). A highly overlapping set of genes was associated with a greater baseline to 2 yr. increase in ppFEV1 and included those related to immune response and inflammation (CXCR5, IL1RL1, NFKBID, S100A10), actin cytoskeleton regulation (RHOA, CFL1, ARHGAP4, CAPZB), and squamous/cornified epithelium (KRT6A, IVL; Figure 5B and Supplemental Table 10). In both cases, a greater increase in ppFEV1 was associated with higher baseline POU2F3, a transcription factor of the rare cell lineage that includes CFTR-rich ionocytes (31). Genes whose lower baseline expression was associated with a greater increase in ppFEV1 included those related to lipid metabolism (SCP2, INSIG2, CEL) and mitochondrial function (PPA2, PMPCB, ALDH6A1, ALDH5A1, COQ3) for baseline to 6 mo., and those related to lipid metabolism (INSIG2, SLC2A13, CRBN), mitochondrial function (PPA2, COQ3, UQCRFS1P1, NFS1), and proliferation (ACTL6A, CENPE, CENPF, GEN1) for baseline to 2 yr. (Supplemental Figure 6B and Supplemental Table 10). WGCNA-based analysis identified a significant (P < 0.05) correlation for the black (cell-mediated immunity) and green (cellular metabolism) modules (Table 3). These data suggest that baseline expression that reflects increased inflammation may be associated with increased ppFEV1 after initiating ETI.
Host transcriptomic responses to ETI are accompanied by changes in the sinonasal microbiome. Substantial airway microbiome changes have been documented in response to ETI therapy, including a drastic decrease in, but not eradication of, common CF airway pathogens (6–13). To assess changes upon ETI initiation, we characterized viral and microbial species in the nasal brush biopsies. A computational approach to identify viral sequences in whole transcriptome RNA sequencing data detected viral reads above threshold in only a few samples. Rhinovirus C was detected in baseline samples from 3 participants (Supplemental Table 11). None of the 6 mo. or 2 yr. samples were positive for any respiratory virus. The microbiome of the inferior turbinate was assessed by 16S ribosomal RNA amplification and sequencing using genomic DNA copurified with the RNA from the brush biopsies. Amplification was successful for all samples. Mean qPCR copy number as a metric of overall microbial abundance decreased substantially over the time course, although it did not reach statistical significance (Figure 6A).
 Figure 6
Figure 6Sinonasal microbiome changes in response to ETI therapy. (A) Microbial abundance indicated by 16S rRNA copy number trends toward decreasing during the time course. Data are shown excluding 2 yr. outliers. Left: Box-and-whisker plot with 1-way ANOVA followed by Tukey’s post hoc test. Right: Individual data points throughout time course overlaid with per–time point mean. (B) Relative abundance among the top 25 sequenced taxa indicates a significant decrease in Pseudomonas (left) and an increase in Moraxella (right) during the time course. Significance assessed with 1-way ANOVA followed by Tukey’s post hoc test.
Due to the limited amount of material obtained from the nasal brushes, taxon-level sequencing was successful for only 15 baseline, thirteen 6 mo., and thirteen 2 yr. samples comprising 7 complete time courses, 11 baseline–6 mo. pairs, seven 6 mo.–2 yr. pairs, and 7 baseline–2 yr. pairs. Staphylococcus, Moraxella, Pseudomonas, Corynebacterium, and Propionibacterium were the most abundant within the top 25 detected taxa (Supplemental Table 12). Detected taxa included both those that contain species that are considered to be a part of the nasal commensal microbiota (Corynebacterium, Dolosigranulum, Anaerococcus) and those that contain CF-associated species (Staphylococcus, Pseudomonas, Streptococcus, Prevotella). Importantly, the relative abundance of Pseudomonas decreased significantly over the time course (Figure 6B). This may indicate a marked reduction in Pseudomonas aeruginosa, the dominant CF airway pathogen in adults in response to ETI, which has been documented by other studies (6–13). Interestingly, the relative abundance of Moraxella was very high in some 2 yr. samples (Figure 6B). Changes in the relative abundance of other taxa or in microbial diversity over the time course were not significant (Supplemental Figure 7, A and B). Importantly, we failed to detect increased viral or bacterial pathogens at 2 yr., suggesting that a rebound in pathogens after 6 mo. of ETI therapy is likely not the main driver of the resurgence of host inflammatory gene expression described above (Figure 1E). The two 2 yr. transcriptional outlier samples contained no detectable viral species, although one showed a high abundance of bacterial taxa but not a higher relative abundance of Pseudomonas (Supplemental Figure 7, C and D). Thus, we cannot confirm that an ongoing infection is the cause of the outlier status. These samples may represent a stronger return to baseline inflammation that would require a richer sample set to fully characterize.
Primary sinonasal airway epithelial cell cultures derived from donors after ETI therapy have mostly improved structure and function. CF basal stem cells are characterized by an “inflammatory memory” such that basal cell populations isolated from the CF airways retain functional defects and compositional changes in in vitro culture (32–35). This stem cell dysfunction is considered to be an important manifestation and a driver of epithelial remodeling. To determine whether the transcriptional changes described above in fact manifest over time in a less remodeled epithelium after initiating ETI therapy, basal stem cells were isolated from nasal brush biopsies at baseline and 6 mo. after ETI (Figure 1A) and differentiated in primary culture. Due to COVID-19 pandemic–related research restrictions that went into effect shortly after starting the 6 mo. sample collection, matched pairs of baseline and 6 mo. samples were only available from a limited number of participants to generate primary air-liquid interface airway epithelial cell cultures (ALIs). No significant differences were observed between the baseline and 6 mo. samples in the total cell counts per brush (1.46 × 106 ± 0.22 × 106 at baseline vs. 1.51 × 106 ± 0.18 × 106 at 6 mo., P = 0.82), the total number of airway epithelial basal cells after expansion (3.81 × 106 ± 0.65 × 106 at baseline vs. 3.49 × 106 ± 0.17 × 106 at 6 mo., P = 0.65), or the number of days required for basal cell expansion in culture (6.17 at baseline vs. 6.55 at 6 mo., P = 0.40). To model in vitro CFTR modulation, both baseline and 6 mo. ALIs were also treated with commercially obtained ETI compounds. Epithelial barrier capacity of mature ALIs measured by transepithelial electrical resistance (TEER) was significantly improved at 6 mo. compared with baseline in 4 out of the 6 cultures (Figure 7A), although measurements remained substantially lower than for ALIs derived from healthy control donors (Supplemental Figure 8A). ETI treatment of ALIs during differentiation had a variable effect on TEER (Supplemental Figure 8B), and thus ETI is likely not sufficient on its own or during such a short timeframe to improve epithelial function. Changes in the extent of mucociliary differentiation were assessed by quantitating ciliated and MUC5AC+ mucous secretory cell numbers. Ciliated cell numbers mirrored the changes in TEER such that ALIs with increased TEER at 6 mo. also contained more ciliated cells (Figure 7, B and D). Mucous secretory cell numbers were either increased or not significantly changed in the 6 mo. cultures compared to baseline (Figure 7, C and D). Overall, these data suggest that the sinonasal basal cell pool from some pwCF returns to a healthier phenotype after ETI with improved mucociliary differentiation, while others retain the dysfunction induced by the CF environment.
 Figure 7
Figure 7Improved epithelial structure and function in primary sinonasal airway epithelial cultures after ETI therapy. (A) Matched baseline and 6 mo. primary sinonasal airway epithelial air-liquid interface (ALI) cultures indicate increased barrier capacity at 6 mo. in most participants as measured by transepithelial electrical resistance (TEER, Ω·cm2). Individual participant graphs show the average of 3 measurements from n = 5 independent cultures. Graphs show box-and-whisker plot with 2-tailed t test. (B) ALIs from matched baseline and 6 mo. samples stained with phalloidin for cell boundaries and labeled with anti-FOXJ1 antibody to mark ciliated cells show improved differentiation at 6 mo. in ALIs with increased TEER. Scale bars: 50 μm. (C) ALIs from matched baseline and 6 mo. samples stained with phalloidin for cell boundaries and labeled with anti-MUC5AC antibody to mark mucous secretory cells show improved differentiation at 6 mo. in some ALIs. Scale bars: 50 μm. (D) Quantitation of FOXJ1- and MUC5AC-positive cells. Individual graphs show the average of at least 3 measurements from n = 3 independent cultures. Graphs show box-and-whisker plots with 2-tailed t test.












