Research ArticleGastroenterology
Open Access | ![]() 10.1172/jci.insight.192953
10.1172/jci.insight.192953
Characteristics of anti–integrin αvβ6 autoantibodies in patients with ulcerative colitis
Ikuhisa Takimoto,1 Masahiro Shiokawa,1 Yoshihiro Nishikawa,1 Takeshi Kuwada,1 Sakiko Ota,1 Darryl Joy C. Juntila,1 Takafumi Yanaidani,1 Kenji Sawada,1 Ayako Hirata,1 Muneji Yasuda,1 Koki Chikugo,1 Risa Nakanishi,1 Masataka Yokode,1 Yuya Muramoto,1 Shimpei Matsumoto,1 Tomoaki Matsumori,1 Tsutomu Chiba,1,2 and Hiroshi Seno1
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Takimoto, I. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Shiokawa, M. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by
Nishikawa, Y.
in:
PubMed
|
Google Scholar
|

1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by
Kuwada, T.
in:
PubMed
|
Google Scholar
|
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Ota, S. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Juntila, D. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Yanaidani, T. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Sawada, K. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Hirata, A. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Yasuda, M. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Chikugo, K. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Nakanishi, R. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Yokode, M. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by
Muramoto, Y.
in:
PubMed
|
Google Scholar
|
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Matsumoto, S. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by
Matsumori, T.
in:
PubMed
|
Google Scholar
|
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Chiba, T. in: PubMed | Google Scholar
1Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, Sakyo-ku, Kyoto, Japan.
2Kansai Electric Power Hospital, Fukushima-ku, Osaka, Japan.
Address correspondence to: Masahiro Shiokawa or Yoshihiro Nishikawa, Department of Gastroenterology and Hepatology, Kyoto University Graduate School of Medicine, 54 Shogoin-Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan. Phone: 81.75.751.4319; Email: machan@kuhp.kyoto-u.ac.jp (MS); nishi328@kuhp.kyoto-u.ac.jp (YN).
Authorship note: MS and YN contributed equally to this work.
Find articles by Seno, H. in: PubMed | Google Scholar
Published January 8, 2026 - More info
JCI Insight. 2026;11(4):e192953. https://doi.org/10.1172/jci.insight.192953.
© 2026 Takimoto et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: March 3, 2025; Accepted: January 2, 2026
-
Results
Anti–integrin αvβ6 antibodies from patients with UC share common CDR3 amino acid sequences. To investigate the properties of anti–integrin αvβ6 autoantibodies from patients with UC, we initially established 15 anti–integrin αvβ6 mAbs using peripheral blood mononuclear cells (PBMCs) or lymph node samples of 7 patients with UC with anti–integrin αvβ6 antibodies. The clinical characteristics of the patients are summarized in Supplemental Table 1 (supplemental material available online with this article; https://doi.org/10.1172/jci.insight.192953DS1). As shown in Figure 1, the 7 patients were given unique identification numbers, P1 to P7, and the obtained mAbs were given unique identification numbers (UC1-1 to UC7-1) along with serial numbers (nos. 1–15). To characterize each mAb, we analyzed complementarity-determining region 3 (CDR3) sequences, which are crucial for antigen specificity, along with variable region gene usage patterns (Figure 1) and the frequency of somatic hypermutations (SHMs) in the V gene region (Supplemental Table 2). SHM frequency was calculated by counting of the number of mutated sequences against the annotated germline sequences of V region using IgBLAST. Using enzyme-linked immunosorbent assay (ELISA), we confirmed that these mAbs reacted with integrin αvβ6 (Figure 2A).
 Figure 1
Figure 1Gene usage and CDR3 amino acid sequence of each mAb. The variable regions of the heavy and light chains of the antibodies were analyzed using IgBLAST. The V, D, and J gene usage, along with the amino acid sequences of the complementarity-determining region 3 (CDR3) for each mAb, is presented.
 Figure 2
Figure 2Evaluation of reactivity of each mAb with integrin αvβ6. (A) The reactivity of each mAb to integrin αvβ6 was evaluated using ELISA in the presence of Mg2+ and Ca2+. All mAbs reacted with integrin αvβ6, albeit with different reactivity. (B) In ELISA in the absence of Mg2+ and Ca2+, all mAbs except UC4 lost reactivity to integrin αvβ6. (C and D) ELISA to estimate the reactivity against integrin αvβ6 of antibodies with a common heavy chain CDR3 amino acid sequence — UC1-1, UC2-3, UC2-4, UC3, and UC6 (carrying the CDR-H1 sequence) (C) and UC1-4, UC2-1, UC2-2, UC5-1, UC5-2, and UC7-1 (carrying the CDR-H2 sequence) (D).
Notably, we identified 2 distinct CDR3 amino acid sequences of the heavy chain that were shared among some of the mAbs obtained from the 7 patients with UC: one was AKVIPRIRGSGKAGIKDYYYGMDV (CDR-H1), encoded by IGHV3-30*18/IGHD3-10*01/IGHJ6*02 (shown in red in Figure 1); and the other was ARDRGFRGDTAMIKGGMDV (CDR-H2), encoded by IGHV1-18*01/IGHD5-18*01/IGHJ6*02 (shown in blue in Figure 1). MAbs no. 1 (UC1-1), no. 7 (UC2-3), no. 8 (UC2-4), no. 9 (UC3), and no. 13 (UC6) shared the first sequence, whereas mAbs no. 4 (UC1-4), no. 5 (UC2-1), no. 6 (UC2-2), no. 11 (UC5-1), no. 12 (UC5-2), and no. 14 (UC7-1) shared the second sequence. Notably, among the mAbs that shared the second sequence in the heavy chain CDR3, UC5-1 had a one-residue variation.
Interestingly, some mAbs had RGD sequences (shown in yellow in Figure 1) in the CDR3 region of the heavy chain. Integrin αvβ6 recognizes RGD sequences of its physiological ligands, including fibronectin and LAP (9). The presence of RGD sequences in the CDR3 regions of these mAbs suggests that they compete with the ligands (e.g., fibronectin and LAP) for binding to integrin αvβ6.
Analysis of CDR3 amino acid lengths and SHM frequencies of the V region gene (Supplemental Table 2) showed that these mAbs had longer CDR3 regions and lower SHM rates in the V region gene than do those reported in healthy individuals (7.47%) (18). Overall SHM frequencies were low; however, mutations were enriched in the CDRs, with higher rates than those in the framework regions (FR) (Supplemental Figure 1A). Amino acid mutations followed a similar distribution (Supplemental Figure 1B).
Most antibodies with CDR-H1 or CDR-H2 carried only 1–3 replacement mutations in the heavy chain CDRs and lacked silent mutations (Supplemental Figure 1C). In the FR regions, antibodies with CDR-H1 tended to show lower replacement/silent ratios, while those with CDR-H2 often showed few replacement mutations and no silent ones (Supplemental Figure 1C). These findings suggest that selective introduction of minimal but functional mutations may be sufficient for antigen binding, and that FR mutations may also contribute to binding in antibodies containing CDR-H2.
SHM levels in light chains varied widely. Some antibodies had heavily mutated CDRs, whereas others retained entirely germline light chains. Notably, several antibodies showed integrin αvβ6 reactivity without any light chain mutations, suggesting that such mutations are not always required for binding.
Taken together, these findings suggest that B cells using germline VH genes with intrinsic autoreactive potential acquire or enhance integrin αvβ6 binding through limited, functionally targeted replacement mutations.
All mAbs except UC4 react against integrin αvβ6 in a cation-dependent manner. Reactivity and affinity of each mAb to integrin αvβ6 were assessed using ELISA and biolayer interferometry (BLI), respectively. To validate results obtained in ELISA, we used the mouse anti–human integrin αvβ6 antibody 10D5, which is known to inhibit both fibronectin and LAP binding (19), as a positive control, and IgG purified from healthy individuals as negative controls. The reactivity of all the mAbs against integrin αvβ6 was confirmed using ELISA (Figure 2A), and the half-maximal effective concentration (EC50) values of the mAbs were calculated (6.30–1,537 ng/mL) (Table 1).
In our previous study, we showed that the serum titer of anti–integrin αvβ6 antibodies in patients with UC increased significantly with the addition of Ca2+ and Mg2+ (4). Therefore, we compared the binding of mAbs to integrin αvβ6 in the presence and absence of Ca2+ and Mg2+. The mAbs except UC4 showed marked loss of reactivity in ELISA in the absence of Ca2+ and Mg2+ (Figure 2B). Ca2+ and Mg2+ are important for integrin heterodimer formation, activation of integrins, and binding to its ligands (20–22). This cation-dependent binding suggests that these antibodies recognize integrin αvβ6 in its active conformation maintained by divalent cations. In other words, these mAbs were directed toward active integrin αvβ6. On the other hand, only UC4 reacted with integrin αvβ6 in a cation-independent manner, indicating that its binding site is different from that of other mAbs.
To investigate differences in reactivity with integrin αvβ6 caused by differences in light chains, we chose mAbs with a common CDR3 amino acid sequence in the heavy chain (Figure 2C: CDR-H1; and Figure 2D: CDR-H2) but with different light chains and analyzed their reactivities. Notably, UC2-3 and UC6 had identical CDR3 amino acid sequences in the light chain, and the light chain CDR3 of UC5-1 and UC7-1 had highly similar amino acid sequences. The other antibodies with common heavy chain CDR3 amino acid sequences had different light chain CDR3 amino acid sequences (UC1-1, UC2-4, and UC3; UC1-4, UC2-1, UC2-2, and UC5-2). UC2-3 and UC6 as well as UC5-1 and UC7-1 showed similar reactivities, consistent with sequence similarity; however, the mAbs with common heavy chain CDR3 amino acid sequences but different light chain amino acid sequences showed different reactivities in ELISA (Figure 2, C and D), suggesting that the light chain amino acid sequence is also important for determining binding specificity to integrin αvβ6.
Next, the affinity of each mAb to integrin αvβ6 was measured using BLI (Table 2 and Supplemental Figure 2A). First, we conducted BLI in the presence of cations in the buffer. Most mAbs had sufficient affinity for integrin αvβ6, similar to the results seen in ELISA; however, some of the mAbs that had shown high EC50 values (UC5-1, UC7-1, and UC7-2) in ELISA showed low affinity in BLI. In particular, UC4 did not bind sufficiently to integrin αvβ6 in BLI. To further characterize the binding properties of UC4, which showed unique cation-independent reactivity in ELISA, we performed BLI measurements using both UC4 and UC1-1 in the absence of cations (Supplemental Figure 2, B and C). We used UC1-1 as a representative cation-dependent antibody. In BLI measurements without cations, neither antibody bound to integrin αvβ6. Notably, UC4 showed no binding in BLI even in the presence of cations, despite showing reactivity in ELISA. The reason for this discrepancy remains to be determined.
 Table 2
Table 2Measurement of the dissociation constant, association rate constant, and dissociation rate constant for the association of each mAb with integrin αvβ6 using biolayer interferometry
Effect of mAbs on integrin αvβ6–fibronectin and integrin αvβ6–LAP binding. To investigate the function of each mAb, we examined blocking activities of the mAbs on integrin αvβ6–fibronectin and integrin αvβ6–LAP binding. Both fibronectin and LAP bind to the RGD binding site of integrin αvβ6 via their RGD motifs (9). In a solid-phase binding assay, 8 of the 15 mAbs firmly blocked integrin αvβ6–fibronectin binding in a dose-dependent manner (Figure 3A). The calculated half-maximal inhibitory concentration (IC50) values are shown in Table 3. However, some mAbs with high EC50 values (UC4, UC5-1, UC7-1, and UC7-2) showed very limited blocking activity. UC2-2, UC5-2, and UC6 did not have sufficient blocking activity to reach a plateau within the measured concentration range, and the IC50 could not be calculated. Notably, mAbs with common heavy chain CDR3 amino acid sequences showed varying degrees of blocking activity (Figure 3, B and C). Only marginal inhibition of integrin αvβ6–LAP binding was observed for the patient-derived mAbs (Figure 3D), in contrast to the clear blocking of integrin αvβ6–fibronectin binding. To better understand this differential blocking effect, we compared the affinities of fibronectin and LAP to integrin αvβ6 using BLI (Supplemental Figure 3, A and B, and Supplemental Table 3). Fibronectin showed measurable binding kinetics (dissociation constant [KD] = 47.18 nM), whereas LAP demonstrated extremely stable binding with negligible dissociation (dissociation rate constant [Koff] < 1.0 × 10–7 1/s); this indicated a substantially higher affinity to integrin αvβ6. The control antibody 10D5 demonstrated much higher affinity for integrin αvβ6 (Supplemental Figure 3C) and was able to effectively block LAP binding. These results suggest that the affinity of patient-derived mAbs for integrin αvβ6 is not sufficient to overcome the strong binding of LAP, thereby limiting their ability to inhibit integrin αvβ6–LAP interaction.
 Figure 3
Figure 3Blocking of integrin αvβ6–fibronectin binding or integrin αvβ6–LAP binding by each mAb in ELISA. (A) Blocking activity of each mAb on integrin αvβ6–fibronectin binding was evaluated using a solid-phase integrin αvβ6 binding assay. MAbs with low EC50 blocked integrin αvβ6–fibronectin binding in a concentration-dependent manner. MAbs with high EC50 concentrations (UC4, UC5-1, UC7-1, and UC7-2) showed little blocking activity. UC2-2, UC5-2, and UC6 did not have sufficient blocking activity to reach a plateau within the measured concentration range. (B and C) Blocking activity on integrin αvβ6–fibronectin binding was compared between antibodies with a common amino acid sequence in the heavy chain CDR3 — UC1-1, UC2-3, UC2-4, UC3, and UC6 (carrying the CDR-H1 sequence) (B) and UC1-4, UC2-1, UC2-2, UC5-1, UC5-2, and UC7-1 (carrying the CDR-H2 sequence) (C). (D) Blocking activity of each mAb against integrin αvβ6–LAP binding was evaluated using the same method. (E and F) Effect of RGDS peptides (E) and control RGES peptides (F) at various concentrations on the binding of mAbs to integrin αvβ6. LAP, latency-associated protein.
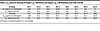 Table 3
Table 3IC50 values for blocking of integrin αvβ6–fibronectin and integrin αvβ6–LAP binding by each mAb in ELISA
Integrin αvβ6 binds to the RGD sequence in its ligands (9). Based on our results and those of a previous study (4), we hypothesized that the mAbs interact with the RGD binding site in integrin αvβ6. Therefore, we evaluated whether RGDS peptides could inhibit the binding of each of the studied mAbs with integrin αvβ6. In line with a previous report (4), RGDS peptides inhibited the binding of all mAbs, except that of UC4, to integrin αvβ6 in a dose-dependent manner (Figure 3E), whereas control RGES peptides did not show such effects (Figure 3F). The binding of UC4 with integrin αvβ6, which showed reactivity with integrin αvβ6 in the absence of cations in ELISA, was inhibited by neither RGDS nor RGES, indicating that this antibody reacts with integrin αvβ6 in an RGD-independent manner. The other mAbs were suggested to be critically dependent on the RGD binding site for binding to integrin αvβ6.
Cross-reactivity of anti–integrin αvβ6 antibodies with integrin αvβ3. Previously, we reported that sera of certain patients with UC contained antibodies against both integrin αvβ6 and αvβ3, with anti-αvβ3 antibody titers being consistently lower than those of anti-αvβ6 antibodies (4). However, it remained unclear whether anti–integrin αvβ3 antibodies existed independently or whether anti–integrin αvβ6 antibodies cross-reacted with integrin αvβ3. Therefore, using ELISA, we tested whether each of the mAbs used in this study reacts with integrin αvβ1, αvβ3, αvβ5, and αvβ8, all of which have αv chains and recognize RGD peptides in their physiological ligands (9) (Figure 4). UC4 showed almost identical reactivity to all integrins used for testing. Therefore, this antibody might react with either the αv chain or a common region of the β chain, such as the β tail domain. UC5-1 and UC5-2 showed reactivity not only to integrin αvβ6 but also to integrin αvβ3. Other mAbs reacted slightly to integrin αvβ3 at high concentrations and had no notable reaction to other integrins. These results suggested that anti–integrin αvβ6 antibodies potentially cross-react with integrin αvβ3.
 Figure 4
Figure 4Cross-reaction of mAbs to other integrins. Using ELISA, each mAb was tested to evaluate whether it reacted with integrin αvβ1, αvβ3, αvβ5, and αvβ8.
IgG and mAbs derived from patients with UC exhibit similar blocking activity on integrin αvβ6–fibronectin/LAP binding. We previously reported that IgG derived from patients with UC blocked integrin αvβ6–fibronectin binding (4); however, we had not tested whether it blocks integrin αvβ6–LAP binding. Using the IgG derived from 42 patients with UC who were part of the training group of our previous study and IgG from 8 control patients (4), we evaluated anti–integrin αvβ6 IgG titer of patients with UC, and then evaluated blocking activity of these IgG samples on integrin αvβ6–fibronectin and –LAP binding (Figure 5, A–C, and Supplemental Table 4).
 Figure 5
Figure 5Evaluation of anti–integrin αvβ6 antibodies using IgG from patients with UC. (A) Presence of anti–integrin αvβ6 antibodies in IgG derived from patients with UC. (B and C) Inhibitory effect of patient-derived IgG on integrin αvβ6–fibronectin binding (B) and integrin αvβ6–LAP binding (C). (D) Correlation of percentage inhibition of integrin αvβ6–fibronectin binding (B) and integrin αvβ6–LAP binding with antibody titers (fibronectin: R2 = 0.6463, P < 0.0001; LAP: R2 = 0.5775, P < 0.0001).
Consistent with our previous results (4), anti–integrin αvβ6 IgG titers were positive in 100% of cases (42 of 42) and showed a substantial inhibitory effect on integrin αvβ6–fibronectin binding in 76.2% of cases (32 of 42). Further, 23.8% cases (10 of 42) showed a substantial inhibitory effect on integrin αvβ6–LAP binding. Both inhibitory effects were correlated with antibody titers (Figure 5D). The patient-derived IgG samples showed blocking activity similar to that of the previously evaluated mAbs, based on their ability to inhibit integrin αvβ6–fibronectin and integrin αvβ6–LAP binding. In both cases, inhibition of integrin αvβ6–fibronectin binding was stronger than that of integrin αvβ6–LAP binding. This similarity suggests that these mAbs capture key features of anti–integrin αvβ6 antibodies in patients with UC.
In contrast to the mAbs, a small proportion of IgG samples from patients exhibited a strong inhibitory effect on integrin αvβ6–LAP binding. It is possible that these individuals possess high-affinity anti–integrin αvβ antibodies, comparable to the 10D5 antibody. However, given that the majority of IgG samples showed limited or no inhibition of integrin αvβ6–LAP binding, such high-affinity antibodies appear to be relatively uncommon and are unlikely to constitute the predominant anti–integrin αvβ6 antibody population in patients with UC.