Research ArticleDermatologyInflammation
Open Access | ![]() 10.1172/jci.insight.179875
10.1172/jci.insight.179875
Tape strip expression profiling of juvenile dermatomyositis skin reveals mitochondrial dysfunction contributing to disease endotype
Jessica L. Turnier,1,2 Sarah M.H. Vandenbergen,1 Madison E. McClune,1 Christine Goudsmit,1 Sophia Matossian,1 Meredith Riebschleger,1 Nadine Saad,1 Jacqueline A. Madison,1,2 Smriti Mohan,1 Johann E. Gudjonsson,2,3 Lam C. Tsoi,3,4,5 Celine C. Berthier,6 and J. Michelle Kahlenberg2,3
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by
Turnier, J.
in:
PubMed
|
Google Scholar
|

1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by Vandenbergen, S. in: PubMed | Google Scholar
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by
McClune, M.
in:
PubMed
|
Google Scholar
|
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by Goudsmit, C. in: PubMed | Google Scholar
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by Matossian, S. in: PubMed | Google Scholar
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by Riebschleger, M. in: PubMed | Google Scholar
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by Saad, N. in: PubMed | Google Scholar
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by
Madison, J.
in:
PubMed
|
Google Scholar
|
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by Mohan, S. in: PubMed | Google Scholar
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by
Gudjonsson, J.
in:
PubMed
|
Google Scholar
|
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by
Tsoi, L.
in:
PubMed
|
Google Scholar
|
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by
Berthier, C.
in:
PubMed
|
Google Scholar
|
1Division of Pediatric Rheumatology, Department of Pediatrics;
2Division of Rheumatology, Department of Internal Medicine;
3Department of Dermatology;
4Department of Computational Medicine and Bioinformatics;
5Department of Biostatistics; and
6Division of Nephrology, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA.
Address correspondence to: Jessica Turnier, Department of Pediatrics, C.S. Mott Children’s Hospital, 2800 Plymouth Road, NCRC Building 20, Room 1842, Ann Arbor, Michigan, 48109, USA. Phone: 440.728.4033; Email: turnierj@med.umich.edu.
Authorship note: SMHV and MEM contributed equally to this work. CCB and JMK contributed equally to this work. CCB and JMK have been designated as co–last authors.
Find articles by
Kahlenberg, J.
in:
PubMed
|
Google Scholar
|
Published March 13, 2025 - More info
JCI Insight. 2025;10(8):e179875. https://doi.org/10.1172/jci.insight.179875.
© 2025 Turnier et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
-
Results
JDM cohort clinical characteristics at enrollment visit. There were 28 JDM and 20 healthy control (CTL) patients in this study. All patients had sampling of nonlesional (NL) skin. Within our JDM cohort, lesional (L) skin was also sampled if rash was present. In the JDM cohort, 17/28 (60.7%) had both a L and NL skin sample collected, and 11/28 (39.3%) had only a NL skin sample collected (Table 1, Table 2, and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.179875DS1). The mean age for JDM diagnosis was 9.0 years for patients with both L and NL skin samples and 8.2 years for JDM patients with NL skin samples only. At the time of tape stripping, the mean age for JDM patients with both L and NL skin sampled (13.3 years) did not differ significantly from JDM patients with NL skin sampling only (11.0 years). Our JDM cohort was predominantly female (23/28, 82.1%) and White (20/28, 71.4%).
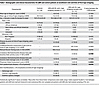 Table 1
Table 1Demographic and clinical characteristics for JDM and control patients at enrollment visit and time of first tape stripping
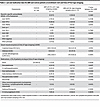 Table 2
Table 2Lab and medication data for JDM and control patients at enrollment visit and time of first tape stripping
We had 2 treatment-naive patients in our cohort, both in the group of patients with L and NL skin samples collected. There was no significant difference in disease duration between the 2 patient groups (4.3 years for JDM with L and NL skin sampling and 2.7 years for NL sampling only). There were more patients with skin-predominant disease in the NL only relative to the group of patients having both L and NL skin samples (5/11 or 45.4% relative to 1/17 or 5.8%). Our JDM cohort had overall low skin disease activity scores. Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) activity scores were higher in patients having both L and NL skin sampling, with a mean Cutaneous Dermatomyositis Disease Area and Severity Index activity score of 7.1 in the L and NL skin group relative to 1.5 in the NL skin only group. Muscle disease activity scores, as assessed by Manual Muscle Testing (MMT-8) and Childhood Myositis Assessment Scale (CMAS), were similar in both cohorts. Physician’s global assessment of disease activity (PGA) scores were slightly higher in the group of patients having both L and NL skin samples (3.2 relative to 1.8). There was not a significant difference in testing positive for presence of an MSA between the L and NL skin group and the NL skin only group (8/15 or 53.3% relative to 3/10 or 30.0%), and TIF1γ was the most represented MSA in the cohort (4/15 or 26.7%). Of note, 2/17 patients in the L and NL skin group and 1/11 patients in the NL skin only group had an unknown MSA status and were not included in these percentages.
Transcriptome analysis of JDM lesional skin highlights interferon and immune activation signatures. To account for patients who had multiple samples from the same tissue type (different time points, NL and/or L), we first combined all samples of a specific tissue type from each patient before differential expression analysis (Supplemental Figure 1). Upon comparison of all JDM L relative to CTL skin samples, we identified 982 differentially expressed genes (DEGs), including 929 upregulated and 53 downregulated genes (FDR < 0.10; Supplemental Table 2). Interferon signaling was the top upregulated pathway (P value < 0.0001), with the top 10 upregulated pathways also including role of pattern recognition receptors in recognition of bacteria and viruses (P value = 0.0014), Th1 and Th2 activation (P = 0.0028), IL13 signaling (P = 0.0036), IL17 signaling (P = 0.0048), and TREM1 signaling (P = 0.01) (Figure 1A, Supplemental Figure 2, and Supplemental Table 3). Central nodes in literature-based network analysis for lesional JDM skin included ISG15, ICAM1, and CSF1, as well as genes involved in the interferon response, immune cell trafficking, and monocyte and macrophage survival (Figure 1B). The top predicted upstream regulators in lesional skin included IRF1 (IPA Z score = 3.95, P = 2.07 × 10–7) and SOCS1 (IPA Z score = –3.37, P = 1.57 × 10–9), which are both genes involved in the regulation of IFN signaling (Supplemental Table 4).
 Figure 1
Figure 1Biological signatures identified in JDM L and NL skin compared with CTL. (A) The left panel displays the top 10 biological pathways (P value < 0.05) regulated in JDM L (n = 17) and NL skin (n = 28) compared with CTL skin (n = 20); the right panel displays selected genes from relevant pathways. Canonical pathway P values were computed by the Ingenuity Pathway Analysis (IPA; QIAGEN) software. nNOS, neuronal nitric oxide synthase. (B) Literature-based network analysis of shared and unique genes in JDM L and NL compared with control skin.
Transcriptome analysis of JDM NL skin is distinct from L skin and exhibits predominant upregulation of metabolic signaling pathways. Interestingly, JDM NL skin demonstrated a higher number of DEGs (n = 4,467, FDR < 0.10) in relation to CTL skin than was seen in L skin comparisons. JDM NL skin displayed 4,138 upregulated and 329 downregulated genes compared with CTL (Figure 1B and Supplemental Table 2). Pathway analysis demonstrated a striking difference compared with JDM lesional skin, with regulation of genes involved in nNOS signaling in skeletal muscle cells (P value = 0.002), neurovascular coupling signaling (P = 0.003), phagosome formation (P = 0.005), and calcium signaling (P = 0.006) (Figure 1A, Supplemental Figure 2, and Supplemental Table 3). In NL skin, EPOR, which encodes the erythropoietin receptor, and SMAD3, which functions in TGFB1 signaling, were among the nodes identified on network analysis (Figure 1B). The top predicted activated upstream regulators expressed in NL skin were the transcription factors HNF1A (Z score = 4.4, P value = 0.0139), which is involved in regulation of glucose metabolism, and ARNT2 (Z score = 4.3, P value = 0.0361), which can participate in regulation of transcriptional response to hypoxia (Supplemental Table 4).
JDM L and NL skin demonstrate shared activation of innate immune signaling pathways. Direct comparison of JDM L with NL skin demonstrated only 10 DEGs (FDR < 0.10), all downregulated in JDM L skin, indicating that the transcriptome of JDM NL skin appears more similar to JDM L skin rather than CTL skin (Supplemental Table 2). Interestingly, 2 of these genes were LOR and FLG2, both related to epidermal barrier function and cornified envelope formation, which have been previously described as downregulated in systemic lupus erythematosus L skin (26). There were 537 DEGs in JDM L overlapping with the NL skin when compared with CTL (Supplemental Table 2 and Figure 1B), and top upregulated pathways in this gene set in common between L and NL skin included role of pattern recognition receptors (P = 0.0026), inflammasome pathway (P = 0.0129), IL13 signaling (P = 0.0191), and IL17 signaling (P = 0.0245) (Supplemental Table 3). Literature-based network analysis of those 537 overlapping DEGs in JDM L and NL skin compared with CTL highlighted increased mRNA expression of BGLAP, encoding osteocalcin, a bone matrix protein potentially involved in calcinosis, as well as CASP1, which can activate IL1B and IL18 in the proper context (Figure 1B). We also identified SOCS1, which is involved in negative regulation of cytokines. The heatmap displayed in Figure 1A and Supplemental Figure 2 highlights some of the upregulated genes in key pathways unique to each JDM L and NL skin and in common between JDM L and NL skin relative to CTL.
Tape stripping and full-thickness skin biopsies share a common interferon expression signature in L skin and alteration in cellular and metabolic signaling pathways in NL skin. To understand the extent to which expression signatures acquired via tape strips reflect the whole skin tissue transcriptome, we compared the JDM tape stripping RNA-Seq transcriptional profile with the full-thickness FFPE skin biopsy microarray expression profile that we previously published (27). The JDM L tape stripping RNA-Seq and full-thickness skin biopsy microarray expression data comparison showed a prominent interferon-stimulated gene expression signature, verifying that tape stripping can effectively recover signatures reflecting the immune dysregulation occurring in inflamed skin. There were a total of 23 genes commonly upregulated in expression datasets from both lesional tape stripping and skin biopsy samples (Figure 2A), including TRIM22, IFI30, USP18, CSF1, IFI6, CYTH4, TRAJ23, HLA-DPA1, HLA-F, IFI16, IFI27, IFIT1, IRF1, MX1, PI3, PLSCR1, SAMD9, IFIH1, LAPTM5, OASL, RSAD2, ISG15, and ADGRE5. A 23-gene signature score generated using these 23 genes showed that this signature was also higher in JDM NL skin tape stripping samples, indicating the detectable presence of an interferon signature even in JDM NL skin but at a lower level than seen in L skin (Figure 2B and Supplemental Figure 3). When comparing JDM NL skin datasets, we noted 100 common DEGs between tape stripping and skin biopsies, with 74 upregulated and 26 downregulated genes (Supplemental Table 2; Figure 2, C and D; and Supplemental Figure 3). Calcium signaling, nNOS signaling in skeletal muscle cells, and mitochondrial biogenesis were among the top represented pathways (P value < 0.05; Supplemental Table 3). Of note, CALM2 and NCOR1 from the mitochondrial biogenesis pathway were both downregulated in JDM NL relative to CTL skin, indicating possible dysregulation in mitochondrial homeostasis in JDM skin even in the absence of clinical inflammation.
 Figure 2
Figure 2Comparison of tape stripping with full-thickness skin biopsy expression signatures. (A) JDM lesional skin. FDR was extracted from the Limma analysis (B) A 23-gene signature from overlap genes in L expression datasets. Data are presented as mean ± SD. Unpaired 2-tailed parametric Student’s t test with Benjamini-Hochberg correction was used for the comparison between 2 groups. (C) JDM NL skin. FDR was extracted from the Limma analysis. (D) A 100-gene signature from overlap genes in NL expression datasets. Data are presented as mean ± SD. Unpaired 2-tailed parametric Student’s t test with Benjamini-Hochberg correction was used for the comparison between 2 groups. RNA-Seq: n = 20 CTL, n = 17 JDM_L, n = 28 JDM_NL; microarrays: n = 8 CTL, n = 9 JDM_L, n = 6 JDM_NL.
Unsupervised hierarchical clustering identifies a JDM molecular subgroup with an expression signature characterized by mitochondrial dysfunction. Unsupervised hierarchical clustering from all the CTL and JDM skin samples identified 2 JDM subgroups with distinct tape stripping expression profiles (corresponding to 65 samples or 28 patients in cluster 1 and 12 samples or 8 patients in cluster 2) (Supplemental Figure 4, A and B). Clustering of enrollment tape stripping samples only (1 sample/individual) produced similar results (Supplemental Figure 4C). We therefore included all tape stripping samples per individual in the analysis. The 6,773 DEGs (FDR < 0.01, absolute log2 fold-change ≥ 1.0; Supplemental Table 2) distinguishing the 2 subgroups represented pathways involving mitochondrial dysfunction, sirtuin signaling, oxidative phosphorylation, protein ubiquitination, and senescence (P value < 0.0001) (Figure 3A and Supplemental Table 3). JDM subgroup 2 demonstrated higher skin-directed interferon, mitochondrial dysfunction, angiogenesis, and innate immune expression scores in skin compared with JDM subgroup 1 and healthy CTL patients (Figure 3B). Upon comparing clinical disease characteristics between subgroups, the 2 subgroups did not separate by L/NL skin status or disease duration or activity (Table 3, Supplemental Table 5, and Supplemental Figure 4). However, patients represented in subgroup 2 were more likely to still be on steroids after similar disease duration, indicating a higher frequency of treatment-refractory disease (Table 4 and Supplemental Table 5), suggesting that tape stripping expression signatures may hold potential to aid in clinical stratification of JDM patients with a different disease subtype that could be more refractory to standard treatment. Of note, the 2 patients in our cohort with treatment-naive samples were split between subgroups, supporting that our findings are not entirely driven by treatment effect. Pathway-based expression scores in skin also did not associate with steroid dose (Supplemental Figure 5).
 Figure 3
Figure 3Unsupervised hierarchical clustering of skin expression data identifies subgroups of patients with JDM. (A) Heatmap of selected DEGs representing pathways reflecting 2 JDM skin subgroups. (B) JDM subgroup 2 demonstrated higher skin-directed interferon, mitochondrial dysfunction, angiogenesis, and innate immune expression scores in skin compared with subgroup 1 and healthy controls. (C) The NFE2L2 signature score was higher in subgroup 2 compared with subgroup 1 and positively associated with the skin-directed interferon score. Unpaired 2-tailed parametric Student’s t test with Benjamini-Hochberg correction was used for the comparison between 2 groups. Cluster 1: n = 65 total samples (n = 45 JDM_NL and 20 JDM_L) corresponding to n = 28 patients; cluster 2: n = 12 total samples (n = 10 JDM_NL and 2 JDM_L) corresponding to n = 8 patients.
NFE2L2 is the top upstream regulator in the JDM treatment-refractory molecular subgroup. NFE2L2 (log2 fold-change = 1.26, FDR < 0.0001), a transcription factor involved in cytoprotective response to oxidative stress and innate immune signaling, was the top upstream regulator activated in JDM subgroup 2 (IPA Z score = 10.74, P value = 6.1 × 10–14) (Supplemental Table 4). There were 221 genes downstream of NFE2L2, from which 72 were involved in at least 1 of the following pathways: mitochondrial dysfunction, protein ubiquitination, sirtuin signaling, unfolded protein response, myelination signaling, and autophagy (in the top 15 pathways from the 6,773 genes regulated in JDM subgroup 2 compared with subgroup 1, Supplemental Table 3 and Supplemental Table 6). A total of 71 of the 72 genes were upregulated in subgroup 2 compared with subgroup 1 and were used to generate an NFE2L2 signature score (Supplemental Table 6). The defined NFE2L2 signature score was higher in subgroup 2 and positively associated with the skin-directed interferon score (P = 0.6427, P < 0.0001) (Figure 3C), indicating an association of NFE2L2 and interferon in JDM pathophysiology.
Validation of JDM molecular subgroup differentiated by mitochondrial dysfunction using independent JDM cohort microarray expression dataset from full-thickness skin biopsies. To verify our findings from tape stripping expression data, we performed unsupervised hierarchical clustering of our previously published microarray data set from FFPE full-thickness skin biopsies of an independent JDM cohort (n = 15) (27) (Supplemental Figure 6). Within this independent JDM skin biopsy cohort, we also identified 2 JDM subgroups with distinct expression profiles, including n = 9 in biopsy subgroup 1 and n = 6 in biopsy subgroup 2 (Supplemental Figure 7). Similar to our JDM tape stripping expression dataset, we identified biopsy subgroup 2 as being characterized by dysregulation in the mitochondrial dysfunction pathway (Figure 4A) and a higher mitochondrial dysfunction signature score (Figure 4B). There were 2,575 DEGs in JDM biopsy subgroup 2 compared with subgroup 1 (FDR < 0.01, absolute log2 fold-change ≥ 1.0) (Figure 4A and Supplemental Table 2). A total of 1,160 of these genes were also regulated in the JDM tape stripping subgroup 2 compared with the JDM tape stripping subgroup 1, with 1,146 regulated in the same direction (all upregulated) and representing protein ubiquitination (P = 4.25 × 10–20), mitochondrial dysfunction (P = 2.68 × 10–9), and oxidative phosphorylation (P = 9.32 × 10–11) among other regulated pathways (Figure 4C, Supplemental Table 2, and Supplemental Table 3). Most of the lesional skin biopsy samples clustered in biopsy subgroup 2, with only 3 in subgroup 1. All NL skin biopsy samples clustered in biopsy subgroup 1. Three patients with JDM naive to systemic treatment at the time of biopsy clustered in biopsy subgroup 2 and only 1 in biopsy subgroup 1.
 Figure 4
Figure 4Validation of a JDM subgroup expression signature using a JDM skin biopsy microarray dataset. (A) Heatmap of the DEGs representing selected pathways commonly activated between the 2 identified JDM subgroups in both skin tape stripping and biopsy. (B) Subgroup 2 demonstrated higher mitochondrial/oxidative phosphorylation dysfunction expression scores in skin compared with subgroup 1 and healthy controls. (C) Comparison of JDM skin expression signatures from tape stripping and full-thickness skin biopsies between the 2 identified subgroups. Unpaired 2-tailed parametric Student’s t test with Benjamini-Hochberg correction was used for the comparison between 2 groups. Cluster 1 RNA-Seq: n = 45 JDM_NL and 20 JDM_L samples corresponding to n = 28 patients; cluster 2 RNA-Seq: n = 10 JDM_NL and 2 JDM_L samples corresponding to n = 8 patients; cluster 1 microarrays: n = 6 CTL, 6 JDM_NL and 3 JDM_L samples/patients; cluster 2 microarrays: n = 0 JDM_NL and 6 JDM_L samples/patients.
Skin as compared with whole blood expression signature more effectively highlights a JDM disease endotype. As whole blood samples were also collected at the time of tape strip sampling if clinical labs were drawn, we evaluated if/how the expression scores from the pathways described above in the skin were reflected in the blood. Based on the 2 identified skin subgroups, we also observed higher skin-directed interferon, angiogenesis, and innate immune scores in the blood from skin subgroup 2 as compared with skin subgroup 1 and CTL (Figure 5A). The identification of a distinct biologic signature not only from skin but also from blood suggests that expression signatures from skin may have the potential to reflect systemic disease. Interestingly, while mitochondrial dysfunction scores from blood were higher in both of our JDM skin subgroups relative to CTL, they were not higher in skin subgroup 2 relative to subgroup 1 (Figure 5A, second panel), which suggests that the blood mitochondrial signature alone could not differentiate the subgroups (Supplemental Figure 7). Thus, the finding of a mitochondrial dysfunction expression signature identifying subgroup 2 was unique to skin (Figure 3, A and B). Similarly, the other pathway scores in blood did not highlight biological differences from the skin-derived subgroup 2 as effectively as seen in skin (Figure 5A compared with Figure 3B).
 Figure 5
Figure 5Whole blood as compared with skin transcriptional profile to define subgroups. (A) Blood expression scores for pathways of interest. Patients with both NL and L skin samples that were split between the 2 defined molecular subgroups are represented by unfilled circles. The blood samples were assigned to subgroup 2 for analysis. (B) Top 10 canonical pathways from the DEGs in L skin from JDM patients with lower Manual Muscle Testing scores (MMT-8 ≤ 146) compared with controls (FDR < 0.10). Unpaired 2-tailed parametric Student’s t test with Benjamini-Hochberg correction was used for the comparison between 2 groups. CTL: n = 6; cluster 1: n = 34 JDM samples; cluster 2: n = 9 JDM samples. Canonical pathway P values were computed by the IPA software.
Upon independent unsupervised hierarchical clustering of blood expression data, we also identified 2 general clusters, but blood subgroups were not as well defined by a clear biological pattern or unique expression signature (Supplemental Figure 8). Patients from skin-derived subgroups were split between blood subgroups (Supplemental Figures 4 and 8), which suggests that transcriptomic skin signatures derived using tape stripping may be able to differentiate a unique patient subgroup, which is not identified in blood.
Patients with JDM with higher muscle disease activity demonstrate increased interferon and myeloid-derived expression signatures in L skin. We also performed differential expression analysis in both L tape stripping and blood expression datasets after stratifying patients with JDM by median organ-specific clinical disease activity scores for skin, muscle, and global disease activity (CDASI activity score of 7, MMT-8 score of 146, and PGA score of 3, defined by median of all patients in cohort). Interestingly, we identified the highest number of DEGs in the tape stripping expression dataset when comparing JDM disease activity subgroups with CTL after stratifying patients by degree of muscle disease activity (MMT-8 score ≤ 146, n = 1,198 DEGs with FDR < 0.10) as compared with skin (CDASI activity score ≥ 7, n = 20 DEGs with FDR < 0.10) or global disease activity (PGA score ≤ 3, n = 88 DEGs with FDR < 0.10). (Supplemental Table 2). The top upregulated pathways included interferon signaling (P = 1.0 × 10–11), IL10 signaling (P = 7.4 × 10–8), role of hypercytokinemia in the pathogenesis of influenza (P = 4.9 × 10–7), dendritic cell maturation (P = 2.1 × 10–6), Toll-like receptor signaling (P = 9.5 × 10–6), CD40 signaling (P = 1.8 × 10–5), and macrophage classical activation signaling pathway (P = 2.6 × 10–5) (Supplemental Table 3 and Figure 5B). In contrast, there were no DEGs noted in the blood expression dataset when the same groups of patients were compared with CTL, indicating the potential clinical utility and possible higher sensitivity of tissue-specific expression signatures in assessing disease activity.
Tape stripping may capture immune cell signatures and suggests a predominant myeloid cell signature in JDM relative to CTL. Using the CIBERSORTx immune cell enrichment analysis tool on our JDM tape stripping expression dataset, we noted enrichment for immune cell expression signatures (Supplemental Table 7). Transcriptomic signatures reflecting myeloid cell populations were particularly represented in L JDM skin (Figure 6A and Supplemental Figure 9A). Immune cell enrichment scores for dendritic cells and neutrophils were higher in L relative to NL JDM skin (Figure 6, A and B, and Supplemental Figure 9A). JDM NL skin reflected a predominant macrophage expression signature and lower B cell signature relative to healthy CTL patients (Figure 6, A and B, and Supplemental Figure 9A), concordant with the enrichment we noted for innate immune activation pathways even in NL skin. JDM NL as compared with L skin demonstrated more enrichment for plasma cells (Figure 6, A and B, and Supplemental Figure 9A).
 Figure 6
Figure 6Immune cell enrichment analysis in JDM and control skin using CIBERSORTx. (A) Heatmap from each relevant immune cell type relative fraction in CTL (n = 21), NL (n = 55) JDM, and L (n = 22) JDM skin. (B) Graphs illustrating the relative fraction of B cells, plasma cells, macrophages, dendritic cells, and neutrophils as computed by CIBERSORTx in CTL and JDM samples (each dot represents 1 sample). (C) Heatmap from each relevant immune cell type relative fraction in each sample from control skin and samples in each identified skin subgroup. (D) Graphs illustrating the relative fraction of B cells and macrophages as computed by CIBERSORTx in each CTL and JDM subgroup sample (each dot represents 1 sample). CTL: n = 21, JDM_NL: n = 55, JDM_L: n = 22, JDM subgroup 1: n = 65, JDM subgroup 2: n = 12. Unpaired 2-tailed parametric Student’s t test with Benjamini-Hochberg correction was used for the comparison between 2 groups.
Upon analysis of immune cell expression scores in JDM skin subgroups, a lower B cell score was found to differentiate JDM skin-derived subgroup 2 from JDM skin-derived subgroup 1 (Figure 6C, Supplemental Figure 9B, and Figure 6D), highlighting that there may be a contribution from differing immune cell–derived signatures and responses in unique JDM disease endotypes. In comparison with healthy CTL skin, JDM skin subgroup 1 reflected a higher macrophage score (Figure 6C, Supplemental Figure 9B, and Figure 6D).












