Bone biology
Abstract
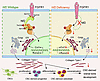
Cranial neural crest cells (CNCs) play a critical role in craniofacial bone morphogenesis, engaging in intricate interactions with various molecular signals to ensure proper development, yet the molecular scaffolds coordinating these processes remain incompletely defined. Here, we identify neurofibromin 2 (Nf2) as a critical regulator to direct CNC-derived skull morphogenesis. Genetic ablation of Nf2 in murine CNCs causes severe craniofacial anomalies, featuring declined proliferation and increased apoptosis in osteoprogenitors, impaired type I collagen biosynthesis and trafficking, and aberrant osteogenic mineralization. Mechanistically, we uncover that Nf2 serves as a molecular linker that individually interacts with FGF receptor 1 (FGFR1) and Akt through spatially segregated phosphor-sites, and structural modeling and mutagenesis identified Ser10 and Thr230 as essential residues, with Thr230 mutation selectively ablating Akt binding while preserving FGFR1 association. Strikingly, Akt inhibition phenocopied Nf2 deficiency, reducing collagen production and Nf2 phosphorylation, whereas phospho-mimetic Nf2 (T230D) rescued CNC-derived osteogenic defects in Nf2-mutant animals. Our findings underscore the physiological significance of Nf2 as a phosphorylation-operated scaffold licensing the FGFR1/AKT axis to regulate collagen type I biogenesis and trafficking, ensuring normal CNC-derived osteogenesis and craniofacial bone development, thus exposing the Nf2/FGFR1/AKT signaling axis as a therapeutic target and promising advancements in treatment of craniofacial anomalies.
Authors
Yuping Huang, Junguang Liao, Panpan Shen, Yiliang He, Fuju Sun, Qi Zhang, Changlin Zheng, Xingen Zhang, Haibo Li, Guiqian Chen
Abstract
Talc pleurodesis is highly effective for preventing recurrence of pneumothorax and pleural effusion, but can be complicated by dissemination, acute lung injury, lead exposure, and foreign body-induced chronic inflammation and pain. Our objective is to develop a safe, biodegradable, contaminant-free particle for pleurodesis. We used mouse models of pneumothorax and malignant pleural effusion to compare the efficacy and safety of pleurodesis with talc and hydroxyapatite microspheres (HAM). Intrapleural instillation of microspheres induced pleural adhesions, fibrosis and symphysis as effectively as talc, and resulted in more durable protection from experimental pneumothorax. HAM and talc both induced an osteoclastogenic, inflammatory and fibrotic response in pleural lavage cells. Intrapleural HAM was resorbed by osteoclast action over 3 months, whereas talc was not cleared. Deletion of the osteoclast effector, CTSK, diminished pleural adhesion formation and fibrosis by talc and HAM, and inhibition of osteoclastogenesis with anti-RANKL antibody delayed HAM clearance. We found no difference in activity level, feeding behavior or lung compliance between particles, but talc induced more persistent pleural inflammation. We conclude that HAM resulted in an osteoclastogenic and fibrogenic pleural response that induced pleurodesis that was more durable than talc with a superior safety profile due in part to osteoclast-mediated particle clearance.
Authors
Yusuke Tanaka, Yuki Takahashi, Yuma Shindo, Lori B. Pitstick, Steven L. Teitelbaum, Wei Zou, Xiangning Wang, Jason Woods, Kathryn A. Wikenheiser-Brokamp, Francis X. McCormack
Abstract
Autophagy is a recycling pathway in which damaged proteins, protein aggregates, and organelles are delivered to lysosomes for degradation. Autophagy insufficiency is thought to contribute to osteoporosis. Accordingly, autophagy elimination from the osteoblast lineage reduces bone formation and bone mass. However, whether increasing autophagy would benefit bone health is unknown. Here, we increased expression of endogenous transcription factor EB gene (Tfeb) in osteoblast lineage cells in vivo via CRISPR activation (TfebCRa mice). Elevated Tfeb stimulated autophagy and lysosomal biogenesis in osteoblasts. TfebCRa mice displayed a robust increase in femoral and vertebral cortical thickness at 4.5 months of age. Increases in cortical thickness was due to increased periosteal bone formation. Tfeb elevation also increased femoral trabecular bone volume. These changes increased bone strength of TfebCRa mice. Female TfebCRa mice displayed a progressive increase in bone mass and at 12 months of age had high cortical thickness and trabecular bone volume. Increased vertebral trabecular bone volume was due to elevated bone formation. Osteoblastic cultures showed that Tfeb elevation increased proliferation and mineral deposition. Overall, these results demonstrate TFEB-driven stimulation of autophagy in osteoblast lineage cells is associated with increased bone formation and strength and may represent an effective approach to combat osteoporosis.
Authors
Alicen James, James A. Hendrixson, Ilham Kadhim, Adriana Marques-Carvalho, Jacob Laster, Julie Crawford, Jeff Thostenson, Visanu Wanchai, Amy Y. Sato, Intawat Nookaew, Jinhu Xiong, Maria Almeida, Melda Onal
Abstract
Parathyroid hormone (PTH) regulates serum calcium and phosphate through its actions in bone and the kidney and is used to increase bone in osteoporosis treatment. In bone, PTH targets osteoblasts and osteocytes to regulate bone remodeling but also bone marrow stromal cells (BMSCs), regulating their differentiation in the osteoblast and/or the adipocyte lineages. PTH exerts its action through the PTH/PTH-related peptide (PTHrP) receptor (PTH1R), a G protein-coupled receptor (GPCR), activating adenylyl cyclase and phospholipase C (PLC). Although the effects of cAMP and PKA are well characterized, little is known about the effects of PLC activation or on the cross-talk between PTH signaling and other pathways. Here, bulk RNA-seq of PTH-treated murine BMSC line (W-20) revealed significant changes in the Hippo pathway. PTH stabilized YAP, a key target of Hippo, by decreasing YAP/LATS1 interaction, YAPS127 phosphorylation and YAP ubiquitination, leading to YAP nuclear translocation and expression of YAP target genes. Similar events occurred in osteocyte cell lines. This occurred via an increase in Src kinase activity: we identified YAPY428 as a key tyrosine residue phosphorylated by Src in response to PTH. Preventing YAP428 phosphorylation led to YAP instability, blocking both osteogenic and adipogenic differentiation of W-20 cells. These results demonstrate active crosstalk between the PTH/PTHrP and the Hippo signaling pathways and reveal that PTH signaling utilizes the PLC-Ca2+-Src tyrosine kinase signaling cascade to influence YAP stability, antagonizing Hippo signaling and favoring stromal cell differentiation. Thus, PTH signaling counteracts the effects of Hippo signaling in BMSCs to favor their differentiation.
Authors
Sara Monaci, Mengrui Wu, Hiroyuki Okada, Kedkanya Mesil, Byeong-Rak Keum, Maisa Monseff Rodrigues da Silva, Clifford J. Rosen, Francesca Gori, Roland Baron
Abstract
Metabolic health is influenced by adipose tissue, and obesity and lipodystrophy are characterized by inflammation and metabolic dysfunction. Whereas obesity and lipodystrophy treatments involve pharmacological approaches and lifestyle changes, these therapies require long-term, repeated dosing, and are not successful for all patients. Gene therapy with targets such as FGF21 and sTGFBR2 provides an alternative approach, specifically in lipodystrophy. Preclinical experiments in mice housed at 22°C are confounded by a mild cold stress not generally experienced by humans, which can negatively affect translation of metabolic therapeutics. In this study, we investigated effects of FGF21/sTGFBR2 combination gene therapy on obese and lipodystrophic mice, and how housing temperature influences therapeutic efficacy. In obese mice, FGF21/sTGFBR2 improved insulin resistance and hyperlipidemia more dramatically at warmer temperatures. In lipodystrophic mice on a high fat diet, combination therapy required adipose tissue to improve insulin resistance at 30°C, whereas FGF21 alone improved insulin resistance at 22°C. Transcriptomic analyses revealed that lipodystrophic mice had upregulated hepatic cell proliferation and fibrosis pathways, and that FGF21 promoted hepatic metabolism. Thus, metabolic dysfunction caused by lipodystrophy is improved by targeting FGF21 and TGFB signaling, but effectiveness in preclinical models may be dependent upon environmental temperature and presence of adipose tissue.
Authors
Jessica N. Maung, Yang Chen, Keegan S. Hoose, Rose E. Adler, Hadla Hariri, Mia J. Dickson, Taryn A. Hetrick, Gabriel A. Ferguson, Rebecca L. Schill, Hiroyuki Mori, Romina M. Uranga, Kenneth T. Lewis, Isabel D. K. Hermsmeyer, Donatella Gilio, Christopher de Solis, Amber Toliver, Noah Davidsohn, Elif A. Oral, Ormond A. MacDougald
Abstract
The aryl hydrocarbon receptor (AhR) is proposed to mediate the frailty-promoting effects of the tryptophan metabolite kynurenine (Kyn), which increases with age in mice and humans. The goal of the current study was to test whether administration of pharmacological AhR inhibitors, BAY2416964 and CH-223191, could abrogate musculoskeletal decline in aging mice. Female C57BL/6 mice (18 months old) were treated with vehicle (VEH) or BAY2416964 (30 mg/kg) via daily oral gavage 5 days/week for 8 weeks. A second AhR antagonist, CH-223191, was administered to 16-month-old male and female C57BL/6 mice via intraperitoneal injections (3.3 mg/kg) 3 days/week for 12 weeks. While grip strength declined over time in VEH-treated mice, BAY2416964 preserved grip strength in part by improving integrity of neuromuscular junctions, an effect replicated during in vitro studies with siRNA against AhR. Cortical bone mass was also greater in BAY2416964- than VEH-treated mice. Similarly, CH-223191 treatment improved cortical bone and showed beneficial effects in skeletal muscle, including reducing oxidative stress as compared to VEH-treated animals. Transcriptomic and proteomic data from BAY2416964-treated mice supported a positive impact of BAY2416964 on molecular targets that affect neuromuscular junction function. Taken together, these data support AhR as a therapeutic target for improving musculoskeletal health during aging.
Authors
Kanglun Yu, Sagar Vyavahare, Dima W. Alhamad, Husam Bensreti, Ling Ruan, Anik Tuladhar, Caihong Dai, Joseph C. Shaver, Alok Tripathi, Kehong Ding, Rafal Pacholczyk, Marion A. Cooley, Roger Zhong, Maribeth H. Johnson, Jie Chen, Wendy B. Bollag, Carlos M. Isales, William D. Hill, Mark W. Hamrick, Sadanand Fulzele, Meghan E. McGee-Lawrence
Abstract
Achondroplasia (ACH) and hypochondroplasia (HCH), the two most common types of dwarfism, are each caused by FGFR3 gain-of-function mutations that result in increased FGFR3 signaling, disrupting chondrogenesis and osteogenesis resulting in disproportionately shortened long bones. In this study, TYRA-300, a potent and selective FGFR3 inhibitor, was evaluated in three genetic contexts: wild-type mice, the Fgfr3Y367C/+ mouse model of ACH, and the Fgfr3N534K/+ mouse model of HCH. In each model, TYRA-300 treatment increased naso-anal length, tibia and femur length. In the two FGFR3-altered models, TYRA-300-induced growth partially restored the disproportionality of long bones. Histologic analysis of the growth plate in Fgfr3Y367C/+ mice revealed that TYRA-300 mechanistically increased both proliferation and differentiation of chondrocytes. Importantly, children with ACH can experience medical complications due to foramen magnum stenosis, and TYRA-300 significantly improved the size and shape of the skull and foramen magnum in Fgfr3Y367C/+ mice. Spinal stenosis is also a frequent complication, and TYRA-300 increased the lumbar vertebrae length and improved the shape of the intervertebral discs in both models. Taken together, these studies demonstrate that the selective FGFR3 inhibitor TYRA-300 led to a significant increase in bone growth in two independent FGFR3-driven preclinical models as well as in wild-type mice.
Authors
Jacqueline H. Starrett, Clara Lemoine, Matthias Guillo, Chantal Fayad, Nabil Kaci, Melissa Neal, Emily Pettitt, Melissandre Pache, Qing Ye, My Chouinard, Eric L. Allen, Geneviève Baujat, Robert L. Hudkins, Michael B. Bober, Todd Harris, Ronald V. Swanson, Laurence Legeai-Mallet
Abstract
Septic arthritis, the most severe joint disease, is frequently caused by Staphylococcus aureus (S. aureus). A substantial proportion of patients with septic arthritis experience poor joint outcomes, often necessitating joint replacement surgery. Here, we show that monocyte depletion confers full protection against bone erosion in a septic arthritis mouse model. In the infected synovium, Ly6Chigh monocytes exhibited increased expression of osteoclastogenesis-related molecules, including CCR2, c-Fms, and RANK. S. aureus lipoproteins induced elevated levels of RANKL, MCSF, and CCL-2 in joints, with synovial fibroblasts identified as the major RANKL producer. Anti-RANKL treatment prevented bone destruction in both local and hematogenous septic arthritis murine models. Importantly, combining anti-RANKL treatment with antibiotics provided robust protection against joint damage. Our results indicate that the infiltration and transformation of monocytes into bone-destructive, osteoclast-like cells are key mechanisms in septic arthritis. Combining anti-RANKL and antibiotic therapy represents a promising therapy against this devastating disease.
Authors
Zhicheng Hu, Meghshree Deshmukh, Anders Jarneborn, Miriam Bollmann, Carmen Corciulo, Pradeep Kumar Kopparapu, Abukar Ali, Mattias N.D. Svensson, Cecilia Engdahl, Rille Pullerits, Majd Mohammad, Tao Jin
Abstract
Mutations in the anoctamin5 (ANO5) gene can lead to musculoskeletal disorders, with monoallelic (autosomal dominant) mutations typically presenting as skeletal abnormalities known as Gnathodiaphyseal dysplasia (GDD). Clinically, GDD is characterized by thickened cortices of long bones and mandibles, narrowed medullary cavities, and increased bone fragility. While autophagy is necessary in regulating bone formation, the specific relationship between ANO5 and autophagy remains poorly understood. In this study, we demonstrated that Ano5 deficiency activates autophagy in mouse cranial osteoblasts (mCOBs), leading to enhanced osteogenic capacity in Ano5-/- mCOBs. The application of 3-Methyladenine (3-MA) and chloroquine (CQ) reversed the excessive osteogenesis observed in Ano5-/- mCOBs. Further analysis revealed that Ano5 deficiency upregulates the expression of ATG9A, and silencing ATG9A significantly reduces both autophagy and osteogenic activity in Ano5-/- mCOBs. Additionally, the AMP-activated protein kinase (AMPK) was found to regulate ATG9A positively, and inhibiting AMPK reduced ATG9A expression, which in turn mitigated excessive osteogenesis of Ano5-/- mCOBs. Moreover, in vivo experiments confirmed that treatment with 3-MA alleviates the bone phenotype abnormalities in Ano5-/- mice. These findings suggest that Ano5 negatively regulates autophagy, contributing to illuminate pathogenesis of GDD. Meanwhile, this research highlights potential therapeutic strategies targeting autophagy to pave the way for the clinical manifestations of GDD.
Authors
Shuai Zhang, Shengnan Wang, Sirui Liu, Xiu Liu, Mingyue Zhang, Huichong Xu, Xiaoyu Wang, Hongyu Li, Ying Hu
Abstract
Intervertebral disc degeneration (IDD) is associated with low back pain, a leading cause of disability worldwide. Fibrosis of nucleus pulposus (NP) is a principal component of IDD, featuring an accumulation of myofibroblast-like cells. Previous study indicated matrix metalloproteinase 12 (MMP12) expression is upregulated in IDD but its role remains largely unexplored. We here showed that TGF-β1 could promote myofibroblast-like differentiation of human NP cells along with an induction of MMP12 expression. Intriguingly, MMP12 knockdown not only ameliorated the myofibroblastic phenotype but also increased chondrogenic marker expression. Transcriptome analysis revealed that the MMP12-mediated acquisition of myofibroblast phenotype was coupled to processes related to fibroblast activation and osteogenesis and pathways mediated by MAPK and Wnt signaling. Injury induced mouse IDD showed NP fibrosis with marked increase of collagen deposition and αSMA-expressing cells. In contrast, MMP12 knockout mice exhibited largely reduced collagen I and III but increased collagen II and aggrecan deposition, indicating an inhibition of NP fibrosis along with an enhanced cartilaginous matrix remodeling. Consistently, an increase of SOX9+/CNMD+ but decrease of αSMA+ NP cells was found in the knockout. Altogether, our findings suggest a pivotal role of MMP12 in myofibroblast generation, thereby regulating NP fibrosis in IDD.
Authors
Yi Sun, Wai Kit Tam, Manyu Zhu, Qiuji Lu, Mengqi Yu, Yuching Hsu, Peng Chen, Peng Zhang, Minmin Lyu, Yongcan Huang, Zhaomin Zheng, Xintao Zhang, Victor Y. Leung
No posts were found with this tag.


