Research ArticleNeuroscienceOphthalmology
Open Access | ![]() 10.1172/jci.insight.192929
10.1172/jci.insight.192929
LRP2 is a potential molecular target for nonsyndromic pathological myopia
Kimberley Delaunay,1 Emilie Picard,1 Patricia Lassiaz,1 Laurent Jonet,1 Vidjea Cannaya,1 José Maria Ruiz-Moreno,2 Kentaro Kojima,3 Henrik Vorum,4,5 Bent Honoré,5,6 Jorge Ruiz-Medrano,2 Lasse Jørgensen Cehofski,4,5 Eric Pussard,7 Renata Kozyraki,8 Alicia Torriglia,1 Olivier Cases,1 and Francine Behar-Cohen1,9,10
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Delaunay, K. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by
Picard, E.
in:
PubMed
|
Google Scholar
|

1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Lassiaz, P. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Jonet, L. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Cannaya, V. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Ruiz-Moreno, J. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by
Kojima, K.
in:
PubMed
|
Google Scholar
|
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Vorum, H. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Honoré, B. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Ruiz-Medrano, J. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Cehofski, L. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Pussard, E. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Kozyraki, R. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Torriglia, A. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Cases, O. in: PubMed | Google Scholar
1Centre de Recherche des Cordeliers, Sorbonne Université, INSERM, Université de Paris Cité, Paris F-75006, France.
2Servicio de Oftalmologia, Hospital universitario Puerta de Hierro-Majahonda, Universidad Castilla La Mancha, Albacete, Spain.
3Department of Ophthalmology, Kyoto Prefectural University of Medicine, Kyoto, Japan.
4Department of Ophthalmology, and
5Department of Clinical Medicine, Aalborg University Hospital, Aalborg, Denmark.
6Department of Biomedicine, Aarhus University, Aarhus, Denmark.
7Department of Genetic and Hormonology, Assistance Publique-Hôpitaux de Paris, Hôpital du Kremlin Bicêtre, Université de Paris-Saclay, Kremlin-Bicetre F-94270, France.
8INSERM U1096, Faculty of Health, University of Rouen, Rouen F-76000, France.
9Assistance Publique-Hôpitaux de Paris, Hôpital Cochin Ophtalmopole, Paris F-75014, France.
10Hôpital Foch, Department of Ophthalmology, Suresnes, France.
Address correspondence to: Francine Behar-Cohen, 15 rue de l’École de Médecine 75006 Paris, France. Email: francine.behar@gmail.com.
Authorship note: RK, AT, OC, and FBC contributed equally to this work.
Find articles by Behar-Cohen, F. in: PubMed | Google Scholar
Published June 24, 2025 - More info
JCI Insight. 2025;10(15):e192929. https://doi.org/10.1172/jci.insight.192929.
© 2025 Delaunay et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: March 4, 2025; Accepted: June 18, 2025

-
Results
Levels of LRP2 are decreased in the vitreous of NSHM eyes with PS. LRP2 levels were evaluated in vitreous samples of patients with HM and PS who underwent pars plana vitrectomy for myopic tractional maculopathy, macular hole, or epiretinal membrane and compared to vitreous from emmetropic patients operated for epiretinal membrane or for intraocular lens luxation. These measurements were carried out on a Japanese population and on a White European population by 2 independent research groups and 2 different analytical methods. Table 1 recapitulates the demographic characteristics and surgical indications.
A proteomic analysis was performed on the vitreous from 15 Japanese patients with NSHM compared to 10 controls. Axial length in the HM group was 29.33 ± 1.94 mm and all patients had PS. Proteomic analysis of the vitreous performed by label-free quantitative liquid chromatography–tandem mass spectrometry identified 332 proteins (Supplemental Data File 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.192929DS1), of which were 73 differentially expressed proteins (DEPs), including LRP2 (fold change = 0.5; P = 0.016) (Figure 1A and Tables 2 and 3). The top upregulated DEPs included apolipoprotein D, TGF-β–induced ig-H3 (TGFBI), complement factor D, and thrombospondin 4. On the other hand, versican core protein, S-arrestin, collagen IX α2 chain, LRP2, and retinol-binding protein 3 (IRBP) were reduced. The most significant biological processes associated with downregulated proteins were linked to nervous system development and negative regulation of proteolysis (Supplemental Figure 1). Molecular functions related to the significantly regulated proteins included reduced LDL binding, calcium ion binding, and chaperone binding, whereas serine-type endopeptidase inhibitor activity and integrin binding were elevated (Supplemental Figure 1). The downregulated proteins were predominantly located in the extracellular region and extracellular space, while upregulated proteins were mostly linked to extracellular matrix (ECM) (Supplemental Figure 1). Abnormal retinal morphology, myopia, and retinal detachment were the most significant phenotypes associated with the DEPs (Figure 1B). The most significant connected protein cluster contained 14 proteins, including LRP2 and its ligands clusterin (CLU) and apolipoprotein E (APOE) (Figure 1C and Supplemental Figure 2A), which were enriched in neurodegenerative disorders (Supplemental Figure 2B). In the White European group of patients, 10 HM patients and 13 emmetropic patients were included. In the HM group, axial length was 29.67 ± 2 mm and all eyes had PS. LRP2 was measured by enzyme-linked immunosorbent assay (ELISA) analysis on vitreous samples. Total protein–normalized LRP2 levels were significantly lower in HM eyes (0.8 ± 0.36 and 0.41 ± 0.21 ng/mL) than in emmetropic eyes (1.7 ± 1.1 and 0.75 ± 0.49 ng/mL) (P = 0.01 and 0.02, respectively) (Figure 1D).
 Figure 1
Figure 1Analysis of proteins in the vitreous of myopic and emmetropic patients. (A) Volcano plot of differentially expressed proteins. The rightmost part of the plot (blue circles) shows the 20 upregulated proteins and the leftmost (red circles) the 53 downregulated proteins. PM, pathological myopia. (B) Monarch analysis (https://monarchinitiative.org/) enrichment of downregulated proteins; only the top 5 terms are displayed. Gene count associated with a particular Monarch term is indicated. (C) Protein-protein interactions (PPIs) using STRING analysis (see Supplemental Methods), displaying the signaling network between the differentially expressed proteins. Nodes in blue circles refer to upregulated proteins, nodes in gray circles refer to downregulated proteins. Markov cluster algorithm analysis indicated the main interactomes. The 9 top interactomes are indicated at the right. (D) Concentrations of LRP2 protein in ng/mL in undiluted vitreous from control (n = 13) and NSHM (n = 10) eyes quantified with ELISA. The graph on the right shows the ratio of LRP2 compared to the total protein content. Statistical analysis was performed using the Mann-Whitney U test. *P < 0.05.
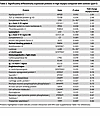 Table 2
Table 2Significantly differentially expressed proteins in high myopia compared with controls (part I)
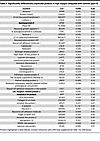 Table 3
Table 3Significantly differentially expressed proteins in high myopia compared with controls (part II)
Pathological features of HM human eyes with PS. Two HM postmortem eyes, with axial lengths of 32 mm and 31 mm for the right and left eye, and a PS, were studied. The premortem spectral domain optical coherence tomography (SD-OCT) images of the right pseudophakic HM eye revealed posterior incurvation of the PS with thinning of the retina and the choroid (Figure 2, A and B; black arrows), complicated by myopic tractional maculopathy (MTM) with foveoschisis (Figure 2, A and B; stars and white stars). The SD-OCT images of the left eye were of low quality due to corneal opacification. On the postmortem enucleated donor eyeball, the PS with scleral thinning was clearly visible (Figure 2C). Peripapillary atrophy was visible on macroscopic pictures of the open eyeball (Figure 2, D and E), and the PS was located temporal to the fovea (Figure 2, D and E; black arrow). A cross section at the level of the PS (Figure 2G) showed disorganization of the retinal layers and cysts (arrows) and extreme retinal thinning at the bottom of the PS (Figure 2G; double arrow) compared with emmetropic retina (Figure 2F). RPE cells outside the PS showed clear disorganization (Figure 2, H–J), with mislocalization of melanosomes and aggregations of pigments. More precise cellular disorders were characterized using markers of different cell types on immunohistochemistry of emmetropic and HM human retina cryosections (Figure 3). In the control retina, glial Müller (RMG) cells identified by glutamine synthetase (GS) expression extended their processes from the inner limiting membrane to the outer limiting membrane (Figure 3, A and B). In the HM retina, RMG processes were disorganized and lost their alignments within the thinned layers (Figure 3C). The RMG processes organized around cysts (Figure 3D) and invaded the bottom of the PS that was covered by RMG cells (Figure 3E). The neuron-specific marker tubulin β3 labeled the nerve fiber layer, the ganglion cells (RGCs) and their extensions, and amacrine cells (Figure 3, F and G). In the HM retina, a severe thinning of the nerve fiber layer (Figure 3H) was observed together with a reduction in RGCs (Figure 3, H and I). Blue and green cones (Figure 3, J and K), labeled by opsins, showed significant disorganization in HM retina with greater loss of blue cones (Figure 3, L and M). Finally, the astrocyte-specific marker glial fibrillary acidic protein (GFAP), which stains astrocytes and the end feet of RMG cells in the emmetropic retina (Figure 3, N and O), showed a dense layer at the surface of HM retinas, which could correspond to the vitreoretinal tractional membrane seen on SD-OCT (Figure 2, A and B) but the RMG invading the PS did not express GFAP. The retina of the HM eye exhibited loss of RGCs and cones, as well as thinning of all retinal layers, and cystic degeneration accompanied by RMG cell disorganization. Extreme degeneration was present in the PS area, which was filled by RMG cells.
 Figure 2
Figure 2Ophthalmologic imaging and histological analysis of the right eye of an HM donor. (A and B) Premortem SD-OCT scans of the right eye with inferior cross section (A) and macular cross section (B), showing the posterior staphyloma (PS) (white arrows) and tractional maculopathy (yellow stars). The myopic eye displayed an atrophy of the macula delineated by small arrows and the posterior incurvation indicated the uncomplicated PS. (C) Gross morphology of the HM right enucleated postmortem eye with PS. (D) Macroscopic image of the posterior chamber after dissection showing the PS. (E) Insert box of D focusing on fovea and PS (black arrow). (F) DAPI-stained section of a normal human retina near the macula. (G) DAPI-stained section of the HM retina with a PS (double headed arrow) shows a severe reduction in thickness of all retinal layers, adjacent to the PS. Schisis (white arrow) is observed between the inner nuclear layer (inl) and the outer nuclear layer (onl). A cyst is observed between the RPE and the outer segments layer (asterisk). bv, blood vessel. (H) RPE cells in human retina form a single layer of pigmented cells as observed on light transmission microscopy. Melanosomes (m) have an elongated shape and nuclei (n) are spaced at regular intervals. (I and J) In the HM donor eye, RPE cells are still forming a single layer. RPE cells are abnormally large, their apical domain is reduced, and melanosomes aggregate in macromelanosomes (mm) or macrostructures (ag). Scale bars: 75 μm (F), 120 μm (G), and 5 μm (H–J).
 Figure 3
Figure 3Immunolabeling of retinal cells in the emmetropic and HM retina. (A and B) Glutamine synthetase (GS) labels Müller glial cells in the emmetropic retina, extending radially from the nerve fiber layer (nfl) to the outer nuclear layer (onl). (C and D) In HM retina, GS-positive cells extend in all directions and thicken at the retinal surface. At higher magnification, GS-positive cells surround a cyst in the ganglion cell layer (gcl) (C) and fill the bottom of the staphyloma where almost no retina remains (E). (F and G) Tubulin β3 (Tubb3) is a neuronal marker of retinal ganglion (RGC) and amacrine cells in the emmetropic retina. (H) In HM retina, the number of Tubb3-positive cells and RGCs is reduced. Higher magnification shows Tubb3-positive axons browsing at the surface of the staphyloma. (J and K) Blue (red staining) and green (green staining) opsin markers reveal the distribution of blue and green cones in the emmetropic retina. (L and M) In HM retina, the number of blue cones is reduced (L) and high magnification shows the accumulation of green opsin in abnormally shaped outer segments. (N and O) Glial fibrillary acidic protein (GFAP) labels astrocytes and glial Müller cell endfeet in the emmetropic retina. (P and Q) In the HM retina, the GFAP-positive endfeet are reduced. (Q) At higher magnification, a GFAP-positive membrane lays on the retinal surface but no positive cells are observed in the retina. Scale bars: 200 μm (A, F, J, and N), 80 μm (B, G, and O), 20 μm (K), 250 μm (C, H, L, and P), and 60 μm (D, E, I, M, and Q).
Decreased LRP2 in human HM with PS retina is associated with severe RPE disorganization similar to Foxg1-Cre-Lrp2fl/fl mouse RPE. In emmetropic human neural retinas, LRP2 was expressed in RMG cells, colocalized with GS (Figure 4, A and B). In the HM neural retinas, significant disorganization of the retinal layers was evident at the margin of the staphyloma, where RMG cells spanning through layers and the subretinal space expressed low levels of LRP2 (Figure 4, C and D). In the RPE cells, LRP2 expression was decreased in the HM eye, as seen in both transversal sections from the left eye (Figure 4E) and flat mounts of the right eye (Figure 4, F–H). In emmetropic RPE, LRP2 was located at the apical membrane and in vesicles (Figure 4E; arrowhead) but also showed diffuse labeling underneath Bruch’s membrane, in the most inner part of the choroid where it stained the pillars of the choriocapillaris (Figure 4E). In RPE flat mounts of the emmetropic control eye, LRP2 was localized in punctiform submembrane clusters at the apical and lateral borders of the RPE cells (Figure 4, F and G), at the cell membrane (Figure 4G), and in clathrin-positive vesicles (Figure 4H). On transversal sections of HM RPE cells (Figure 4I), LRP2 was faintly detected (Figure 4, J–L) at the cell membrane (Figure 4K) and in very few clathrin-positive vesicles (Figure 4L), with staining of LRP2 aggregated at the apical side (Figure 4I; arrowhead) without any signal underneath Bruch’s membrane (Figure 4I). Along with LRP2 reduction, a parallel reduction in clathrin-positive vesicles (Figure 4L) was observed. In HM eyes with PS, LRP2 was thus decreased in the vitreous, in retina, and in the RPE. As opposed to the regular arrangement of RPE cells in emmetropic human eye (Figure 5A), in HM eyes, the RPE displayed irregular organization, cell size dispersity (Figure 5B), and doubling or breaks of the cell junctions (Figure 5, C and D). The RPE from Foxg1-Cre-Lrp2fl/fl mice was studied to explore the impact of suppressed LRP2 expression on the RPE morphology. As opposed to control WT mice (Figure 5, E–G) that showed each regular octagonal RPE cell surrounded by 8 regular hexagonal cells, the transgenic mouse displayed an irregularly arranged RPE with dramatic changes in cellular size and shape mostly posterior to the equator (Figure 5, H and I). Membrane ZO-1 staining in tight junctions was lost and replaced by diffuse cytoplasmic staining (Figure 5J). At the periphery, areas with normal-sized RPE cells showed actin stress fibers (Figure 5J). Automatic quantification (Figure 5K) revealed that RPE cell size increased in the middle and peripheral retina (Figure 5, L and M) and that cell density decreased mostly posterior to the equator (Figure 5N).
 Figure 4
Figure 4LRP2 immunolabeling in the emmetropic and HM retina. (A and B) In the emmetropic neural retina, LRP2 is expressed around RGCs and along GS-positive glial Müller cells. (C and D) In the HM neural retina, LRP2 expression is greatly reduced in GS-positive cells, which surround cysts (stars). (E) In transversal sections of RPE cells, LRP2 is located at the apical pole (white arrowheads), in intracellular vesicles, and at the basal pole of the RPE, and in the pillars of the choriocapillaris. (F and G) In an RPE flat-mounted preparation from the left emmetropic eye, LRP2 is distributed in cytoplasmic vesicles (F) along apical and lateral membranes (G) and most LRP2-positive vesicles are also positive for clathrin. (I) In transversal section of the HM RPE, LRP2 expression is greatly diminished and absent in the choriocapillaris. (J–L) In RPE flat mounted from the HM eye, LRP2 distribution is sparse and diffuse, and does not colocalize with clathrin (L) that is also diminished. Scale bars: 200 μm (A–D), 10 μm (E and I), and 50 μm (F, G, and J–L).
 Figure 5
Figure 5Organization of the RPE is altered in HM eye, similar to the RPE in Lrp2-cKO mice. (A) Phalloidin staining reveals the geometric paving pattern of RPE in the emmetropic eye. (B–D) In HM eye, RPE geometric paving pattern is lost as most cells increase in size. Bicellular junctions display secondary actin arcs (C, arrows) and cell junctions are disorganized (D arrows). (E and F) Phalloidin staining on RPE flat mount of WT control mouse shows the regular pattern of RPE cells. (G) Zonula occludens 1 (ZO-1) follows the distribution of phalloidin in the apical pole. (H and I) In Lrp2-cKO RPE, the pseudogeometric paving pattern is lost and is replaced by a tangle of cells whose surface has increased. (J) ZO-1 is redistributed in the cytoplasm of abnormally shaped RPE cells. (K) Digital overlay reconstruction of control and Lrp2-cKO RPE indicates the increase size in cells both at the periphery and at the intermediate level of the retina in Lrp2-cKO RPE. (L) Cell area in μm2, (M) cell density in number of cells/mm2, and (N) distribution of cells by sizes in the intermediate and peripheral retina of control compared to Lrp2-cKO mice. Values represent the mean of cell size average of each sample (n = 7) per genotype ± SEM. Mann-Whitney U test. ***P < 0.001. Scale bars: 50 μm (A–D, E, and H), 25 μm (F and I), and 75 μm (G and J).
The RPE from individuals with HM and PS, which exhibited a marked decrease in LRP2 expression, displayed morphological changes analogous to those observed in Lrp2-cKO mice.
Transcriptional consequences of LRP2 silencing in iRPE cells. To decipher the consequence of LRP2 downregulation, we used RNA interference to silence LRP2 mRNA (siLRP2) in human induced pluripotent stem cell–derived RPE (iRPE) cells. We first evaluated the ability of differentiated iRPE to express LRP2 and other endocytosis markers. As expected, LRP2 was localized at the cell membrane from the apical to the basolateral side and in subapical vesicles (Figure 6A), colocalizing at the apical pole with the receptor-mediated endocytic protein clathrin (Figure 6B), partially with the early endocytic marker EEA1, (Figure 6C) and with LAMP1, a membrane protein principally located in lysosomes (Figure 6D). Bulk transcriptomes showed that siLRP2 reduced LRP2 mRNA expression by 40% after 48 hours, which was confirmed by quantitative PCR (qPCR) (Figure 7A) and by Western blot (Figure 7B). Principal component analysis (PCA) showed the overall variation among samples, with a clear separation in the first 3 components between siLRP2-treated and scrambled siRNA–treated iRPE cells (Supplemental Figure 3). We identified 118 differentially expressed transcripts between siLRP2 iRPE and scramble iRPE, including 75 downregulated and 42 upregulated transcripts (Figure 7C and Supplemental Data File 2). Shortened lists of differentially expressed genes (DEGs) with the greatest fold change are provided in Tables 4 and 5. Complete lists of DEGs with additional information about their roles in eye physiology are provided in Supplemental Tables 1 and 2. Along with LRP2, we identified several other RPE-specific apical membrane DEGs that were downregulated in siLRP2 iRPE, while upregulated DEGs were involved in the control of the circadian clock, cell cycle, and in the regulation of TGF-β activity. The results of the Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis demonstrated that DEGs were mainly enriched in “retinol metabolism” (downregulation) and in “ECM receptor interaction” (upregulation) (Supplemental Figure 4A and Supplemental Data File 3). The Gene Ontology–based (GO-based) enrichment analysis performed under the 4 categories Biological Processes, Cellular Components, Molecular Functions, and Gene Sets (Supplemental Figure 4, B–E show the topmost enriched GO terms; Supplemental Data File 3 has the complete list) identified the downregulation of genes related to molecular transmembrane transporter activity, fat-soluble vitamin metabolic process (related to retinoic acid and vitamin D), ion homeostasis (Supplemental Figure 4, B and D), and apical and basal plasma membranes (Supplemental Figure 4, C and E, and Supplemental Figure 5A). The upregulated DEGs were enriched in GO terms related to ECM and cytoskeleton arrangement (Supplemental Figure 4, C and D). Using Reactome pathway analysis (Supplemental Data File 3), the top downregulated proteins included the canonical retinoid cycle in vision and solute carrier–mediated transmembrane transport (Supplemental Figure 5A), while the top upregulated DEGs included kinesins, collagen biosynthesis, and modifying enzymes (Supplemental Figure 5A). Human Phenotype Ontology enrichment analysis (Supplemental Data File 3) identified chorioretinal atrophy, RPE atrophy, and progressive night blindness (Supplemental Figure 5B). Enrichment analysis with LRP2 as a common factor (Supplemental Data File 4) showed enriched GO terms related to the apical plasma membrane, solute transmembrane transporters, fat-soluble vitamin metabolic processes, sensory perception, and steroid metabolic processes (Figure 7D). Upregulated enriched GO terms were related to the cytoskeleton and collagen-containing ECM (Supplemental Data File 4). Reactome pathway analysis identified 3 downregulated pathways: visual phototransduction, metabolism of steroids, and sensory perception (Figure 7E). Table 6 recapitulates selected GO gene sets and their potential link with myopia and PS development.
 Figure 6
Figure 6LRP2 localization in healthy human iRPE. (A) Consecutive confocal images from an apical to basal stack showing LRP2 accumulation in large vesicular structures in the apical pole (arrows), at lateral membranes, in small cytoplasmic vesicles, and in larger perinuclear vesicles (arrowheads). (B) Consecutive confocal images from an apical to basal stack show that LRP2 colocalized with the major protein of coated pits and apical endocytic vesicles, clathrin (yellow arrow). In the basal part of the cell, LRP2 (green arrow) did not colocalize with clathrin (red arrow). (C) Confocal image showing that LRP2 (green) partially colocalized with the endocytic marker, early endosome antigen 1 (EEA1; red) (yellow arrow). (D) In the basal compartment, LRP2 (green) colocalized with LAMP1 (red), a specific lysosomal marker (yellow arrows). Scale bars: 10 μm (A, C, and D) and 14 μm (B).
 between siLRP2 and scrambled iRPE cells.") Figure 7
Figure 7Differentially expressed genes (DEGs) between siLRP2 and scrambled iRPE cells. (A) Lrp2 mRNA quantification between scrambled (n = 4) and siLRP2 (n = 4) iRPE (*P < 0.05). (B) LRP2 levels as expressed by the ratio LRP2/GAPDH between scrambled (n = 4) and siLRP2 (n = 4) iRPE (**P < 0.01). (C) Volcano plot of DEGs between siLRP2 and scrambled iRPE cells with –log10 of the adjusted P value on the y axis and log2 of the fold change in expression on the x axis. Some genes, including LRP2, are indicated. (D) Category netplot gene enrichment analysis considering only LRP2 as a common factor with 3 GO terms (sensory perception, import across plasma membrane, and steroid metabolic processes) and (E) with 2 Reactome terms (sensory perception, visual transduction and metabolism). Fold change (color codes on each graph) for each gene of the selected GO term is indicated. Analysis was performed using Metascape (https://metascape.org/). Absolute normalized enrichment allowed identifying terms that were upregulated or downregulated using all DEGs.
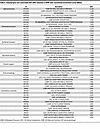 Table 6
Table 6Selected gene sets associated with LRP2 reduction in iRPE cells: normalized enrichment scores (NESs)
Light and cortisol regulate LRP2 expression in human iRPE cells. Light and circadian rhythms are major environmental factors associated with the incidence and the progression of myopia (27) and cortisol secretion, which is under circadian regulation, is modulated by light exposure (28). We assessed the exposure of iRPE cells to an LED system emitting red (631 nm), blue (454 nm), and hot white (3300K) light at a dose of 0.3 J/cm², which was previously shown to be safe for the iRPE (29). After 0.5 and 2 hours of exposure to any of these LEDs, the expression of LRP2 mRNA increased significantly (fold change 2.00 ± 0.16, P = 0.0059 and 1.70 ± 0.08, P = 0.0132) and transiently, returning to baseline at 10 hours (fold change 1.02 ± 0.18, P = 0.9791) (Figure 8A). Exposure to red light was the most effective at increasing LRP2 mRNA at 0.5 and 2 hours (fold change 2.08 ± 0.14, P = 0.0286 and 1.83 ± 0.06, P = 0.0286) (Figure 8A). On immunohistochemistry, 2 hours after illumination, we observed a parallel increase in LRP2 and EEA1 in iRPE cells (Figure 8B).
 Figure 8
Figure 8LRP2 expression and environmental factors. (A) Quantification of LRP2 mRNA by qPCR in iRPE not exposed (ne) or exposed to light (left panel) after 0.5, 2, or 10 hours after illumination. Left panel showing quantification of LRP2 mRNA in iRPE not exposed or exposed to light. Right panel showing quantification of LRP2 mRNA in iRPE not exposed or exposed to red, blue, or white light. Values correspond to the means of 4 independent experiments in duplicate for each condition. Each independent experiment represents the mean of 3 wells. Data are expressed as fold change in gene expression ± SD and were analyzed using the nonparametric Kruskal-Wallis test and Mann-Whitney post hoc test. *P < 0.05; **P < 0.01; #P > 0.05 (not significant). (B) LRP2 and EEA1 expression in iRPE not exposed or exposed to red light. (C) LRP2 mRNA expression in iRPE cultures treated without (ethanol) or cortisol. Values correspond to the mean of 6 experiments in triplicate. Data represent the mean fold change in gene expression ± SEM. Mann-Whitney U test. *P < 0.05. Scale bars: 20 μm (LRP2) and 40 μm (EAA1).
Having previously demonstrated that the glucocorticoid receptor NR3C1 (GR) and the mineralocorticoid receptor NR3C2 (MR) are both expressed and functional in the iRPE cells used in this study (30), we evaluated the effect of cortisol on LRP2 expression. Exposure of iRPE cells to 100 nM cortisol led to a significant decrease in LRP2 mRNA expression after 24 hours (fold change 1.00 ± 0.04 vs. 0.67 ± 0.07; P = 0.0152) (Figure 8C).
Interestingly, light also induced the regulation of the NR3C1 and NR3C2 genes in iRPE cells. Both white and blue light increased the expression level of NR3C1 as compared with non-illuminated cells (fold change 1.1 ± 0.1 vs. 1.5 ± 0.4 [P = 0.01] and vs. 1.8 ± 0.18 [P > 0.0001]), but not red light (Supplemental Figure 6), suggesting that blue light induces RPE cells’ sensitivity to cortisol to a greater extent. NR3C2 expression was increased similarly by red and blue light (fold change 1 ± 0.2 vs. 2 ± 0.6 and 2 ±1.4; P = 0.0001 for both).
Both light exposure and cortisol regulate the expression of LRP2 in iRPE cells but in an opposite manner, suggesting that LRP2 could be one of the molecular links between environmental factors and HM development.









 between siLRP2 and scrambled iRPE cells.")






