Clinical Research and Public HealthClinical ResearchOncology
Open Access | ![]() 10.1172/jci.insight.187073
10.1172/jci.insight.187073
Loss of GATA2 promotes invasion and predicts cancer recurrence and survival in uterine serous carcinoma
Usha S. Polaki,1 Trey E. Gilpin,1 Apoorva T. Patil,1 Emily Chiu,2 Ruth Baker,3 Peng Liu,4,5 Tatiana S. Pavletich,1 Morteza Seifi,6 Paula M. Mañán-Mejías,2 Jordan Morrissey,1 Jenna Port,1 Rene Welch Schwartz,4 Irene M. Ong,4,5,7 Dina El-Rayes,8 Mahmoud A. Khalifa,8 Pei Hui,9 Vanessa L. Horner,1,6 María Virumbrales-Muñoz,2,5,10 Britt K. Erickson,3 Lisa Barroilhet,2 Stephanie M. McGregor,1,5 Emery H. Bresnick,7,11 and Daniel R. Matson1,5,11
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Polaki, U. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Gilpin, T. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Patil, A. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Chiu, E. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Baker, R. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Liu, P. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Pavletich, T. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Seifi, M. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Mañán-Mejías, P. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Morrissey, J. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Port, J. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Welch Schwartz, R. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Ong, I. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by El-Rayes, D. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Khalifa, M. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Hui, P. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Horner, V. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Virumbrales-Muñoz, M. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Erickson, B. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by Barroilhet, L. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by McGregor, S. in: PubMed | Google Scholar
1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by
Bresnick, E.
in:
PubMed
|
Google Scholar
|

1Department of Pathology and Laboratory Medicine and
2Department of Obstetrics and Gynecology, University of Wisconsin-Madison, Madison, Wisconsin USA.
3Department of Obstetrics, Gynecology, and Women’s Health, University of Minnesota, Minneapolis, Minnesota, USA.
4Department of Biostatistics and Medical Informatics and
5Carbone Cancer Center, University of Wisconsin–Madison, Madison, Wisconsin, USA.
6Wisconsin State Laboratory of Hygiene, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin, USA.
7Department of Cell and Regenerative Biology, University of Wisconsin–Madison, Madison, Wisconsin, USA.
8Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota, USA.
9Department of Pathology, Yale University School of Medicine, New Haven, Connecticut, USA.
10Department of Biomedical Engineering and
11Wisconsin Blood Cancer Research Institute, University of Wisconsin–Madison, Madison, Wisconsin, USA.
Address correspondence to: Daniel R. Matson, Room 1770 WIMR, Department of Pathology & Lab Medicine, 1111 Highland Ave., Madison, Wisconsin 53705, USA. Phone: 608.265.4377; Email: drmatson@wisc.edu.
Authorship note: USP and TEG contributed equally to this work.
Find articles by
Matson, D.
in:
PubMed
|
Google Scholar
|
Published April 1, 2025 - More info
JCI Insight. 2025;10(9):e187073. https://doi.org/10.1172/jci.insight.187073.
© 2025 Polaki et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: September 19, 2024; Accepted: March 21, 2025

-
Results
GATA2 IHC predicts recurrence and survival in USC. We first performed GATA2 IHC on a retrospective cohort (UW1 cohort; Supplemental Table 2) of 70 primary USC tumors representing all FIGO stages that were removed at the time of staging hysterectomy, prior to any adjuvant therapy. This cohort was chosen because it had been previously assembled by one of the coauthors for prior studies (5, 16). All patients were diagnosed between 2003 and 2012, and patient follow-up extended through 2019. When compared using univariate and multivariate analyses to 1,710 patients in the SEER (https://seer.cancer.gov/) database diagnosed with USC from 2018 to 2021 and for whom sufficient clinicopathologic metrics were available for analysis, our UW1 cohort had similar patient age at diagnosis, tumor size, FIGO stage, rate of neoadjuvant chemotherapy, and rate of adjuvant chemotherapy (Supplemental Tables 3 and 4). However, our UW1 cohort had a significantly higher percentage of white women and significantly lower rates of adjuvant radiotherapy administration than those reported in SEER. The primary outcomes analyzed were: (a) recurrence free survival, defined as survival without recurrence of USC; (b) cancer-related survival, defined as no death due to USC; and (c) overall survival, defined as no death due to any cause.
For GATA2 IHC, we utilized a mouse anti-GATA2 monoclonal antibody, which we previously demonstrated to be sensitive and specific for mouse and human GATA2 across diverse assays, including Western blot of whole cell lysate and IHC performed on benign and pathologic formalin-fixed paraffin-embedded endometrial biopsies (17, 18). To further confirm antibody specificity, we also performed IHC on the same cohort using a unique and extensively validated anti-GATA2 rabbit polyclonal antibody pool (19–22), which yielded an identical staining pattern as the monoclonal antibody (Supplemental Figure 2). We also previously performed anti-GATA2 immunoprecipitations (IPs) from whole cell lysate using the validated rabbit polyclonal anti-GATA2 antibody and showed that our anti-GATA2 monoclonal specifically recognized GATA2 in the anti-GATA2 IP lane relative to the species and isotype control lane (17). Tumor nuclei were either negative or positive for GATA2 (Figure 1A), which is a pattern of GATA2 expression that we have previously reported in benign and premalignant uterine tissues (18), as well as in myelodysplastic neoplasms and acute leukemias in the bone marrow (17). Because expression of related GATA family members GATA3 and GATA6 has been reported in uterine tissue (23, 24), we also performed GATA3 and GATA6 IHC on our UW1 cohort and confirmed that GATA3 and GATA6 expression did not correlate with GATA2 (Supplemental Figure 3).
 Figure 1
Figure 1GATA2 IHC correlates with low FIGO stage and with cancer-related and overall survival in USC. (A) H&E sections and GATA2 IHC from representative USCs showing spectrum of GATA2 IHC staining. Tumor TMA 210 is an example of a GATA2lo USC, while tumors TMA 180 and TMA 188 are both GATA2hi. H&E and DAB, 600×. Scale bar: 50 μm. (B) Histogram depicting percent GATA2+ tumor nuclei in all 70 cases from UW1 USC cohort. Vertical dotted line indicates 15% cutoff between GATA2lo and GATA2hi USCs. (C–F) Distribution of patient age (C), tumor size (D), FIGO stage (E), and myometrial invasion (F) between GATA2lo and GATA2hi USCs (n = 70). (G) Kaplan-Meier curve depicting cancer-related survival in USCs from UW1 cohort based on GATA2lo or GATA2hi status (n = 64). Analysis in C, D, and F by Student’s t test and analysis in E by Fisher’s exact test. **P < 0.005, ***P < 0.0005.
The percentage of GATA2+ USC tumor nuclei ranged from 0% to 100% (Figure 1B), with an initial inflection point at 15% that we later found clearly separated patients by survival. In selecting a cutoff value for USC GATA2 scoring, we also sought to establish a straightforward GATA2 IHC scoring method amenable to routine surgical pathology practice. Therefore, we established 15% GATA2+ nuclei as a cutoff and assigned all USCs with fewer than or equal to 15% GATA2+ nuclei as GATA2lo, and USCs with greater than 15% GATA2+ tumor nuclei as GATA2hi (Figure 1B).
Patients with GATA2hi USCs were of similar age (Figure 1C) and had tumors that were the same size (Figure 1D) as patients with GATA2lo USCs. However, patients with GATA2hi USC presented at lower FIGO stage (Figure 1E) and showed significantly reduced myometrial invasion (Figure 1F), reduced uterine lymphovascular invasion (LVI), and were more frequently positive for estrogen receptor (ER) and PR compared with GATA2lo USCs (Supplemental Table 5). Patients with GATA2hi USC also had significantly better cancer-related and overall survival compared with patients with GATA2lo USC (Figure 1G and Supplemental Figure 4A), and GATA2 status was independently predictive of overall survival (Supplemental Table 6). We also analyzed GATA2 RNA transcript levels in 2 of the largest publicly available patients with USC cohorts (25, 26) and found no significant predictive value of GATA2 transcript levels for survival, suggesting that RNA transcript levels are less informative than protein-based analyses (Supplemental Figure 4, B–E).
Interestingly, 75% of GATA2hi USCs were from patients with cancer limited to the uterine body at diagnosis, representing FIGO stage I disease. Among the 30 patients in our UW1 cohort with metastatic, FIGO stage III–IV USC, only 3 (10%) had GATA2hi USCs. Therefore, to further elucidate relationships between GATA2 expression and survival in patients with metastatic USC, we scored GATA2 IHC on an additional independent retrospective USC cohort (Yale cohort, n = 36; Supplemental Table 7), which included 28 primary tumors from patients with FIGO Stage III–IV USC (27). We analyzed the GATA2 status in the combined cohort of 58 metastatic USCs (FIGO stage III–IV tumors from the UW1 and Yale cohorts, n = 58; Supplemental Table 8). This revealed no significant relationship between GATA2 status and survival (Supplemental Figure 5). We concluded that GATA2 expression predicts recurrence and survival in USC but only in patients with nonmetastatic USC. Therefore, we focused further analyses on patients diagnosed with USC limited to the uterine body. These patients with FIGO stage I USC represent 50% of new USC diagnoses.
GATA2 status predicts cancer recurrence and survival in FIGO stage I USC. Under FIGO staging criteria (and similar American Joint Committee on Cancer criteria) (28), FIGO stage IA USCs are either noninvasive or invasive but penetrate less than 50% of the uterine myometrium, while FIGO stage IB USCs invade through 50% or more of the myometrium but remain contained within the uterine body. In both cases, there is no evidence of USC outside of the body of the uterus. It is recommended that surgical staging be performed in these patients, to include removal of the uterus, cervix, ovaries, fallopian tubes with lymph node evaluation (using sentinel lymph node removal or complete pelvic and para-aortic lymphadenectomy) and omental biopsy. This identifies microscopic disease and ensures accurate stage assignment.
Our initial UW1 USC cohort included 35 FIGO stage I USCs, and our Yale cohort included 3 FIGO stage I USCs. We augmented these cases by generating 2 new and unique retrospective cohorts representing 89 additional FIGO stage I USCs. The first new cohort consisted of 45 USC tumors derived from FIGO stage IA and IB patients who had been treated at the University of Wisconsin Hospital and Clinics (UW2 cohort, n = 45; Supplemental Table 9). GATA2 IHC and scoring was performed similarly to the UW1 cohort, except whole slides of tumor were stained and scored. For the second new cohort, the anti-GATA2 monoclonal antibody was independently optimized for IHC by the University of Minnesota (UMN) Medical Center (n = 44; Supplemental Table 10) where GATA2 IHC was scored blindly on 44 FIGO stage IA noninvasive USC tumors derived from UMN patients. Scoring at UMN was performed blindly by a senior UMN gynecologic pathologist and a pathology resident who were asked to score the percent GATA2+ tumor nuclei without any additional training. The total pooled FIGO stage I cohort consisted of patients pooled from the UW1, UW2, Yale, and UMN cohorts. This represented 127 total FIGO stage I USCs with matched survival data, a median follow-up period of 3.2 years, and variable amounts of matched clinicopathologic metrics (Table 1 and Supplemental Figure 1).
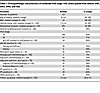 Table 1
Table 1Clinicopathologic characteristics of combined FIGO stage I USC cohort pooled from cohorts UW1, UW2, UMN, and Yale
We found that 33% of FIGO stage I USCs were GATA2hi (Figure 2A). Patients with GATA2hi FIGO stage I USCs were the same age at diagnosis (Figure 2B) and had the same BMI (Supplemental Table 11) as patients with FIGO stage I GATA2lo USCs. In addition, GATA2hi FIGO stage I USCs had the same tumor size (Figure 2C), the same incidence of uterine LVI and positive pelvic washings, and the same expression of ER, PR, and Erb-B2 Receptor Tyrosine Kinase 2 (Her2) compared with GATA2lo USCs (Supplemental Table 11). GATA2hi tumors also had the same degree of myometrial invasion as GATA2lo USCs (Figure 2D). We attribute this to the fact that 67% of our FIGO stage I cohort is composed of FIGO stage IA noninvasive USCs, which by definition have no measurable myometrial invasion. Patients with GATA2hi FIGO stage I USCs showed significantly better recurrence free (P = 0.01) and cancer-related 5-year survival (P = 0.04), and trended toward better overall survival (P = 0.06) compared with GATA2lo USCs (Figure 2, E–G, and Supplemental Table 12). Notably, the 33% of FIGO stage I patients with GATA2hi USCs showed 100% recurrence free and cancer-related survival.
 Figure 2
Figure 2GATA2 IHC predicts recurrence and survival in FIGO stage I USC. (A) Histogram depicting percent GATA2+ tumor nuclei in combined FIGO stage I USC cohort (n = 127). These patients were pooled from UW1, UW2, Yale, and UMN cohorts. Vertical dotted line indicates 15% cutoff between GATA2lo and GATA2hi USCs. (B–D) Spectrum of patient age (n = 127) (B), tumor size at staging hysterectomy (n = 124) (C), and myometrial invasion (n = 121) (D) in FIGO stage I GATA2lo and GATA2hi USCs as analyzed by Student’s t test. (E–G) Kaplan-Meier curves depicting recurrence free (n = 123) (E), cancer-related (n = 78) (F), and overall survival (n = 121) (G) in FIGO stage I–II USCs based on GATA2lo or GATA2hi status. *P < 0.05.
GATA2 depletion promotes invasion in patient-derived USC cells. We hypothesized that GATA2 expression suppressed USC invasion and metastatic spread, and we sought to test whether GATA2 plays a mechanistic role in USC biology or if GATA2 levels simply correlate with aggressive USC behavior. USC pathogenesis occurs in a stepwise manner, in which foundational inactivation of TP53 is followed by acquisition of chromosomal instability (CIN), leading to a “copy number high” state that promotes USC invasion and metastasis through unclear mechanisms (25). Since 93% of USCs in our UW1 cohort were TP53 inactivated (Supplemental Table 2) despite tumors having GATA2 expression ranging from 0% to 100%, it indicated that GATA2 loss did not occur prior to foundational TP53 inactivation. To determine whether GATA2 loss occurs before or after acquisition of CIN, we performed CIN FISH on our UW1 USC cohort and found no significant difference in CIN between GATA2hi and GATA2lo USCs (Figure 3A), supporting a model whereby GATA2 loss occurs after TP53 inactivation and acquisition of CIN, and therefore represents a more proximal molecular readout of USC invasion and metastatic potential.
 Figure 3
Figure 3GATA2 depletion promotes invasion in patient-derived USC cell lines. (A) Level of chromosomal instability in GATA2lo or GATA2hi USCs (n = 60). (B and C) GATA2 is expressed in Ark1 and Ark2 patient-derived USC cells and can be depleted by anti-GATA2 siRNA (B) and doxycline-inducible shRNA (C). (D) Schematic of Matrigel-coated membrane invasion assay. (E) Ability of Ark1 and Ark2 USC cells to invade through Matrigel-coated membranes after GATA2 depletion (n = 11 for Ark1 and n = 7–8 for Ark2). (F) Schematic of hydrogel invasion assay. (G) Ability of Ark1 spheroids to invade into hydrogel after GATA2 depletion (n = 3–5). Analysis in E and G by Student’s t test, with Bonferroni corrections performed for both groups in E. *P < 0.05, ***P < 0.005, ****P < 0.0005. CIN, chromosomal instability; DOX, doxycycline.
Using previously published Ark1 and Ark2 patient-derived USC cell lines (29), we next demonstrated that USC cells express GATA2, which we could deplete in a transient fashion using 2 unique pools of anti-GATA2 siRNA (Figure 3B and Supplemental Figure 6A) or long-term via a doxycycline-inducible anti-GATA2 shRNA (Figure 3C and Supplemental Figure 6B). Since there are 6 GATA factor family members with varying degrees of primary sequence homology and functional overlap (30), we quantified the expression of GATA family members in Ark1 and Ark2 cells using quantitative PCR (qPCR) after GATA2 depletion or scramble control (Supplemental Figure 7A). We detected GATA6 expression in both Ark1 and Ark2 cells and detected GATA3 only in Ark2 cells, neither of which showed significant changes in transcript levels after GATA2 siRNA treatment.
We then tested the effect of GATA2 depletion on USC invasion activity using 2 independent assays. First, we tested the ability of USC cells treated with siGATA2 to traverse Matrigel-covered membranes compared with siScramble controls (Figure 3D). In both Ark1 and Ark2 cells, siGATA2 treatment resulted in a significant increase in invasion compared with siScramble control (Figure 3E). We then sought to complement these findings in a more physiologically relevant system. To this end, we generated 3-dimensional tumor spheroids from Ark1 cells with doxycycline-inducible expression of shGATA2 or shScramble control. Inducible shRNAs were used, as lipofection required for siRNA delivery could not penetrate into tumor spheroids. After addition of doxycycline, spheroids were embedded in natural hydrogel casted within a microfluidic chamber. Over time, tumor invasion was quantified as increased crown area, defined as the invasive front of tumor cells escaping the spheroid core (Figure 3F). Over 72 hours, shGATA2 Ark1 cells showed significantly increased invasion compared with shScramble controls (Figure 3G).
Increased USC invasion was not solely due to increased USC proliferation. While GATA2 depletion did result in increased proliferation in Ark1 USC cells, there was no effect of GATA2 depletion on proliferation in Ark2 USC cells compared with controls (Supplemental Figure 7B). More importantly, GATA2lo patient USC tumors were the same size as their GATA2hi counterparts in patients with USC (Figure 1D and Figure 2C).
GATA2 does not affect USC response to chemotherapy. GATA2 expression has no effect on survival in patients with metastatic (FIGO stages III–IV) USC (Supplemental Figure 5). Because the survival of these patients is largely dependent on response to combination paclitaxel + carboplatin chemotherapy, it implies that GATA2 expression does not affect USC chemotherapy response. However, GATA2 IHC was performed on primary (uterus localized) tumors in our initial analysis, and following staging hysterectomy, it is the response of metastatic USC tumors to chemotherapy that determines patient survival. Therefore, we performed GATA2 IHC on metastatic tumors collected at staging hysterectomy from 22 patients in our UW1 cohort and compared their expression to the primary tumor to determine if USC metastases have different GATA2 levels than the primary tumors (Supplemental Figure 8). Interestingly, the average GATA2 expression in metastatic tumors was equal to the primary tumor in 8 cases (36%), less than the primary tumor in 9 cases (41%), and greater than the primary tumor in 5 cases (23%) (Supplemental Figure 8A). GATA2 expression varied even in different metastatic tumors from the same patient collected at different anatomic locations (Supplemental Figure 8B). Overall, in 77% of cases, metastatic USC tumors have equal to or less GATA2 expression than the primary tumor, but there is substantial variability in GATA2 expression in USC metastases. To test if altered GATA2 expression affected sensitivity to chemotherapy, we depleted GATA2 in Ark1 and Ark2 USC cells, treated them with paclitaxel, carboplatin, or combination paclitaxel + carboplatin, and analyzed cell survival after 96 hours compared with scramble controls (Supplemental Figure 9). This confirmed that GATA2 levels have no effect on USC sensitivity to chemotherapy, in line with USC survival data in FIGO stage III–IV patients (Supplemental Figure 5).
GATA2 multi-omics identifies SIN3B as a USC GATA2 target gene. Since GATA2 is a transcription factor that functions through binding to genomic GATA nucleotide motifs and activating transcription of nearby target genes, we next sought to identify GATA2 networks and gene targets that may normally suppress USC invasion. To this end, RNA-Seq of Ark1 cells performed after GATA2 depletion revealed 8,500 differentially expressed genes (DEGs) (Figure 4A), and gene ontology (GO) term analysis of the top 500 most significantly upregulated and top 500 most significantly downregulated DEGs revealed downregulation of basement membrane reorganization and upregulation of protein localization to the cell cortex, integrin-mediated signaling pathways, and muscle cell migration, respectively (Supplemental Figure 10). We complemented RNA-level analyses by performing anti-GATA2 ChIP-Seq also in Ark1 cells, which revealed 16,816 genomic peaks of GATA2 occupancy, 64% (10,692 peaks) of which were in either proximal gene promoters or introns (Figure 4B). As expected, peaks of GATA2 occupancy were most enriched in GATA consensus nucleotide motifs but also showed significant enrichment for consensus binding motifs corresponding to TAL BHLH Transcription Factor 1 (TAL1), MDS1 and EVI1 Complex Locus (MECOM), Fos Proto-Oncogene (FOS), Jun Proto-Oncogene (JUN), and Cut Like Homeobox 1 (CUX1) (Figure 4C).
 Figure 4
Figure 4GATA2 multi-omics identifies SIN3B as a GATA2 target that regulates USC invasion. (A) Volcano plot of RNA-Seq data showing RNA expression changes after GATA2 depletion in Ark1 USC cells. (B) Number of genome wide GATA2 ChIP-Seq peaks that fall within different gene regulatory regions. (C) Transcription factor motifs enriched within regions of GATA2 ChIP-Seq occupancy. (D) Approach to identifying candidate GATA2-regulated genes in USC. (E) SIN3B locus with superimposed GATA2 ChIP-Seq occupancy. (F) Western blot depicting SIN3B levels after GATA2 depletion in Ark1 USC cells. (G) Western blot showing SIN3B levels after induction of shSIN3B or shScramble control in Ark1 USC cells. (H) USC invasion through Matrigel-coated membranes following depletion of SIN3B or scramble control (n = 11) as analyzed by Student’s t test with Bonferroni correction. *P < 0.05, **P < 0.005.
To identify putative direct GATA2 gene targets, we cross-referenced the genes in which GATA2 bound proximal promoters and/or introns with the genes whose expression was significantly reduced after GATA2 depletion, which yielded 1,031 genes (Figure 4D). To these, we applied a prioritization scheme that gave greater weight to genes based on degree of reduced expression after GATA2 depletion, size of the associated GATA2 occupancy peak, and published involvement in cancer progression. From this emerged SIN3 transcription regulator family member B (SIN3B), which codes for a conserved scaffold protein and histone deacetylase (HDAC) complex subunit that tethers HDAC activity to sequence-specific transcriptional repressors while also stimulating HDAC activity (31, 32). Reduced SIN3B levels impair cellular differentiation and cell cycle withdrawal in response to oncogenic stimuli (33, 34), and SIN3B is reported to variably promote or suppress tumor invasion depending on cancer type (35–37).
GATA2 occupies the SIN3B proximal promoter in USC cells (Figure 4E), and we confirmed that SIN3B levels are reduced after GATA2 depletion (Figure 4F), supporting a model whereby GATA2 normally promotes SIN3B expression. To test whether SIN3B normally suppresses USC invasion, similarly to GATA2, we generated doxycycline-inducible shSIN3B Ark1 cells (Figure 4G) and found that SIN3B-depleted Ark1 cells had a significantly increased capacity to invade through Matrigel-coated membranes compared with shScramble controls (Figure 4H). Finally, we performed IHC for SIN3B on our UW1 cohort and scored both the percent SIN3B+ tumor nuclei and the intensity of SIN3B staining. However, we found no significant relationship between SIN3B and GATA2 expression (Supplemental Figure 11), indicating that GATA2 regulation of SIN3B may not be generalizable across all USC tumor contexts.
GATA2 IHC predicts FIGO stage I USC recurrence in the absence of adjuvant therapy. If GATA2hi FIGO stage I USCs have a greatly reduced capacity to invade and metastasize, and therefore have very high rates of cure by staging hysterectomy, then adjuvant therapy is unlikely to be critical in reducing the rate of USC recurrence in these patients. Moreover, we showed that GATA2 expression does not affect chemotherapy response, again suggesting that adjuvant therapy should play no role in a GATA2-related survival benefit. If GATA2 IHC could predict USC recurrence, then 33% of patients with FIGO stage I USC with GATA2hi tumors could be spared adjuvant therapy and instead undergo a period of close clinical observation. To evaluate this possibility, we identified 24 patients in our combined cohort who had FIGO stage I USC and received no adjuvant or neoadjuvant chemotherapy, including 14 GATA2hi USCs and 10 GATA2lo USCs (n = 24; Supplemental Table 13). These patients represented all patients with FIGO stage I USC from our UW1, UW2, Yale, and UMN cohorts who had not received adjuvant chemotherapy. Among these chemotherapy naive patients, those with GATA2hi USCs had 100% recurrence free survival, which was significantly better than patients with GATA2lo USCs (Figure 5A). The same patients with GATA2hi USCs also had significantly improved overall survival compared with patients with GATA2lo USCs (Figure 5B).
 Figure 5
Figure 5GATA2 IHC predicts recurrence and overall survival in FIGO stage I USC in the absence of adjuvant chemotherapy. (A and B) Kaplan-Meier curves depicting recurrence-free (n = 24) (A) and overall survival (n = 24) (B) in GATA2lo and GATA2hi patients with FIGO stage I USC who did not receive adjuvant chemotherapy. These patients were pooled from UW1, UW2, Yale, and UMN cohorts. (C) Current NCCN guidelines and proposed GATA2-guided treatment model for FIGO stage I USC. Per the NCCN, patients with FIGO stage IA noninvasive USCs may receive either close observation or adjuvant therapy, whereas patients with invasive FIGO stage I USCs should receive adjuvant therapy. In a proposed GATA2-guided model, patients with GATA2hi FIGO stage I USCs would be offered close observation, while patients with GATA2lo FIGO stage I USC would be offered adjuvant therapy. *P < 0.05. NCCN, National Comprehensive Cancer Network.








