Therapeutics
Abstract
Moderate hyperoxia (30–60% O₂) in premature infants promotes bronchial airway hyperresponsiveness (AHR) via airway smooth muscle (ASM), a key regulator of bronchoconstriction, bronchodilation, and remodeling. Understanding how O2 exposure drives long-term bronchial changes in prematurity is critical for developing therapies for airway disease across the lifespan. Premature lungs have immature antioxidant defenses, potentially due to disrupted mitochondrial dynamics, increasing susceptibility to O2-induced oxidative stress. Thus, mitochondrial homeostasis is highly relevant to ASM dysfunction and airway disease. We propose that hyperoxia in prematurity promotes mitochondrial dysfunction, and that the gasotransmitter hydrogen sulfide (H₂S) mitigates O2-induced mitochondrial damage in developing ASM. Human fetal ASM (fASM) were exposed to moderate hyperoxia to investigate the effects of exogenous H₂S donors (GYY4137, AP39) and stabilization of cystathionine β-synthase (CBS), an H₂S biosynthetic enzyme, on mitochondrial structure and function. Hyperoxia impaired fASM mitochondrial integrity, while H₂S donors in particular, or CBS stabilization attenuated adverse O2 effects on mitochondrial morphology, reactive oxygen species, respiration, calcium regulation, and contractility. These findings highlight the therapeutic potential of H₂S in the premature lung exposed to moderate hyperoxia.
Authors
Colleen M. Bartman, Michael Thompson, Samantha K. Hamrick, Niyati A. Borkar, Daniel Pfeffer-Kleemann, Preetham Ravi, Marta Schiliro, Yak Nak, Christian Vivar Ramon, Li Drake, Y. S. Prakash, Christina Pabelick
Abstract
Radiotherapy is a critical modality in cancer treatment, not only to eradicate cancer cells but also to trigger anti-tumor immunity. Interleukin-21 (IL-21), an immunomodulatory cytokine with potential in cancer therapy, has unexplored synergy with radiotherapy. Our study, leveraging human cancer databases and tissue microarrays, identified a positive correlation between IL-21 and radiotherapy outcomes, particularly in tumor microenvironment (TME) activation. In mouse tumor models, IL-21 combined with radiation significantly enhances TME, boosting CD8+ T cell activation and function, reducing tumor burden, and extending survival. Single-cell transcriptome sequencing revealed that the combination of IL-21 and radiation increased the cytotoxicity of effector and memory CD8+ T cells and prevented their exhaustion. These effects were further validated in humanized mice, where IL-21 combined with radiation reduced A549 tumor growth and enhanced CD8+ T cell function. Post-neoadjuvant radiotherapy samples from patients with esophageal cancer showed a positive correlation between IL-21 levels and CD8+ T cell infiltration. Our findings suggest that IL-21 is a promising adjuvant to radiotherapy, potentially improving the treatment efficacy through TME enhancement. This study provides a foundation for future clinical exploration of IL-21 for enhancing radiotherapy.
Authors
Xinyang Li, Xueqi Xie, Baochao Wei, Xiaozheng Sun, Minxin Chen, Rufei Liu, Qingxu Tao, Yiheng Huang, Qian Wang, Shuangshuang Ma, Ling Wei, Rong Xiao, Zhaoyun Liu, Jinming Yu, Meng Wu, Dawei Chen

Abstract
We recently showed that cell surface translocation of the endoplasmic reticulum–resident protein GRP78, when bound by activated α 2-macroglobulin (α2M*), induces pro-fibrotic responses in glomerular mesangial cells in response to high glucose and regulates activation of the pro-fibrotic cytokine transforming growth factor-β1 (TGF-β1), implicating a pathogenic role in glomerulosclerosis. Interstitial fibrosis, largely mediated by proximal tubular epithelial cells (PTEC) and renal fibroblasts, develops later in kidney disease and correlates with functional decline. Here we investigated whether interstitial fibrosis was mediated by cell surface GRP78 (csGRP78)/α2M*. High glucose and TGF-β1 increased csGRP78 and α2M* in PTEC and renal fibroblasts, and their inhibition prevented fibrotic protein production. Interestingly, for TGF-β1, this depended on inhibition of noncanonical signaling through YAP/TAZ, with Smad3 activation unaffected. In vivo, type 1 diabetic Akita mice overexpressing TGF-β1 were treated with either a neutralizing antibody for csGRP78 (C38) or α2M* (Fα2M) or an inhibitory peptide blocking csGRP78/α2M* interaction, and mice with unilateral ureteral obstruction were treated with Fα2M or inhibitory peptide. Consistently, inhibition by antibody or peptide attenuated fibrosis and pro-fibrotic signaling. These findings show an important role for csGRP78/α2M* in mediating tubulointerstitial fibrosis in both diabetic and nondiabetic kidney disease and support their inhibition as a potential antifibrotic therapeutic intervention.
Authors
Jackie Trink, Ifeanyi Kennedy Nmecha, Katrine Pilely, Renzhong Li, Zi Yang, Sydney Kwiecien, Melissa MacDonald, Bo Gao, Mariam A. Mamai, Chao Lu, Urooj F. Bajwa, Nikhil Uppal, James C. Fredenburgh, Masao Kakoki, Salvatore V. Pizzo, Anthony F. Rullo, Matthew B. Lanktree, Jeffrey I. Weitz, Yaseelan Palarasah, Joan C. Krepinsky
Abstract
Epigenetic macromolecular enzyme complexes tightly regulate gene expression at the chromatin level and have recently been found to colocalize with RNA splicing machinery during active transcription; however, the precise functional consequences of these interactions are uncertain. Here, we identify unique interactions of the CoREST repressor complex (LSD1-HDAC1-CoREST) with components of the RNA splicing machinery and their functional consequences in tumorigenesis. Using mass spectrometry, in vivo binding assays, and cryo-EM we find that CoREST complex-splicing factor interactions are direct and perturbed by the CoREST complex selective inhibitor, corin, leading to extensive changes in RNA splicing in melanoma and other malignancies. Moreover, these corin-induced splicing changes are shown to promote global effects on oncogenic and survival-associated splice variants leading to a tumor-suppressive phenotype. Using machine learning models, MHC IP-MS, and ELISpot assays we identify thousands of neopeptides derived from unannotated splice sites which generate corin-induced splice-neoantigens that are demonstrated to be immunogenic in vitro. Corin is further shown to reactivate the response to immune checkpoint blockade, effectively sensitizing tumors to anti-PD1 immunotherapy. These data position CoREST complex inhibition as a unique therapeutic opportunity which perturbs oncogenic splicing programs while also creating tumor-associated neoantigens that enhance the immunogenicity of current therapeutics.
Authors
Robert J. Fisher, Kihyun Park, Kwangwoon Lee, Katarina Pinjusic, Allison Vanasse, Christina S. Ennis, Parisa Farokh, Scott B. Ficarro, Jarrod A. Marto, Hanjie Jiang, Eunju Nam, Stephanie Stransky, Joseph Duke-Cohan, Melis A. Akinci, Anupa Geethadevi, Eric Raabe, Ana Fiszbein, Shadmehr Demehri, Simone Sidoli, Chad W. Hicks, Derin B. Keskin, Catherine J. Wu, Philip A. Cole, Rhoda M. Alani
Abstract
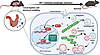
Sepsis contributes substantially to mortality rates worldwide, yet clinical trials that have focused on its underlying pathogenesis have failed to demonstrate benefits. Recently, enhancing self-defense has been regarded as an emerging therapeutic approach. Autophagy is a self-defense mechanism that protects septic mice, but its regulatory factor is still unknown. Moreover, the role of interferon regulatory factor 7 (IRF7) in sepsis has been debated. Here, we showed that Irf7 deficiency increased mortality during polymicrobial sepsis. Furthermore, IRF7 drove macrophages to protect against sepsis. Mechanistically, IRF7 is a transcription factor that upregulates the expression of autophagy-related genes responsible for autophagosome formation and autolysosome maturation, induces autophagic killing of bacteria, and ultimately reduces septic organ injury. Recombinant adeno-associated virus 9–Irf7–mediated IRF7 overexpression promoted the autophagic clearance of pathogens and improved sepsis outcomes, which may be the mechanism underlying the observed improvement in bacterial clearance. These findings provide evidence that IRF7 is the underlying regulatory factor that drives autophagy to eliminate pathogens in macrophages during sepsis. Collectively, IRF7 overexpression represents a potential host-directed therapeutic strategy for preclinical sepsis models, operating independently of antibiotic mechanisms.
Authors
Guiming Chen, Kangxin Li, Haihua Luo, Lianxu Zhao, Yong Jiang
Abstract
Anthracycline chemotherapy, widely used in cancer treatment, poses a significant risk of cardiotoxicity that results in functional decline. Current diagnostic methods poorly predict cardiotoxicity because they do not detect early damage that precedes dysfunction. Positron emission tomography (PET) is well-suited to address this need when coupled with suitable imaging biomarkers. We used PET to evaluate cardiac molecular changes in male C57BL/6J mice exposed to doxorubicin (DOX). These mice initially developed cardiac atrophy, experienced functional deficits within 10 weeks of treatment, and developed cardiac fibrosis by 16 weeks. Elevated cardiac uptake of [68Ga]Ga-FAPI-04, a PET tracer targeting fibroblast activation protein alpha (FAP), was evident by 2 weeks and preceded the onset of functional deficits. Cardiac PET signal correlated with FAP expression and activity as well as other canonical indicators of cardiac remodeling. By contrast, cardiac uptake of [18F]DPA-714 and [18F]MFBG, which target translocator protein 18-kDa (TSPO) and the norepinephrine transporter (NET), respectively, did not differ between the DOX animals and their controls. These findings identify FAP as an early imaging biomarker for DOX-induced cardiac remodeling in males and support the use of FAP PET imaging to detect some cancer patients at risk for treatment-related myocardial damage before cardiac function declines.
Authors
Chul-Hee Lee, Onorina L. Manzo, Luisa Rubinelli, Sebastian E. Carrasco, Sungyun Cho, Thomas M. Jeitner, John W. Babich, Annarita Di Lorenzo, James M. Kelly

Abstract
Butyrate, a microbiome-derived short-chain fatty acid with pleiotropic effects on inflammation and metabolism, has been shown to significantly reduce atherosclerotic lesions, rectify routine metabolic parameters such as low-density lipoprotein cholesterol (LDL-C), and reduce systemic inflammation in murine models of atherosclerosis. However, its foul odor, rapid metabolism in the gut and thus low systemic bioavailability limit its therapeutic effectiveness. Our laboratory has engineered an ester-linked L-serine conjugate to butyrate (SerBut) to mask its taste and odor and to coopt amino acid transporters in the gut to increase its systemic bioavailability, as determined by tissue measurements of free butyrate, produced by hydrolysis of SerBut. In an apolipoprotein E–knockout (ApoE)–/– mouse model of atherosclerosis, SerBut reduced systemic LDL-C, proinflammatory cytokines, and circulating neutrophils. SerBut enhanced inhibition of plaque progression and reduced monocyte accumulation in the aorta compared with sodium butyrate. SerBut suppressed liver injury biomarkers alanine transaminase and aspartate aminotransferase and suppressed steatosis in the liver. SerBut overcomes several barriers to the translation of butyrate and shows superior promise in slowing atherosclerosis and liver injury compared with equidosed sodium butyrate.
Authors
Taryn N. Beckman, Lisa R. Volpatti, Salvador Norton de Matos, Anna J. Slezak, Joseph W. Reda, Ada Weinstock, Leah Ziolkowski, Alex Turk, Erica Budina, Shijie Cao, Gustavo Borjas, Jung Woo Kwon, Orlando deLeon, Kirsten C. Refvik, Abigail L. Lauterbach, Suzana Gomes, Eugene B. Chang, Jeffrey A. Hubbell
Abstract
Glycogen storage disease type Ia (GSD Ia) is caused by a deficiency of glucose-6-phosphatase (G6Pase) in the liver leading to lethal hypoglycemia. Gene therapy with adeno-associated virus (AAV) vectors encoding G6Pase fails to stably treat GSD Ia early in life. We evaluated genome editing in 12 day-old infant mice with GSD Ia using two AAV vectors, one containing Cas9 from Streptococcus pyogenes and a second Donor vector that expresses a guide RNA and a G6PC transgene. Gene therapy with the Donor vector only was compared with genome editing using both Donor and CRISPR vectors. Treatment with genome editing (total vector dose 0.2 to 2E+13 vector genomes/kg) and bezafibrate (to stimulate autophagy) was efficacious as assessed by hypoglycemia prevention and the frequency of transgene integration, which correlated with improved survival. This therapy achieved 5.9% chromosomal transgene integration through homology directed repair, which surpassed a threshold to prevent long-term hepatic complications. No integration was detected in absence of the CRISPR vector. Importantly for safety, CRISPR vector genomes were depleted, and no intact, integrated CRISPR genomes were detected by long-read sequencing. Thus, genome editing warrants further development as a potentially stable treatment for human infants with GSD Ia.
Authors
Benjamin Arnson, Ekaterina Ilich, Troy von Beck, Songtao Li, Elizabeth D. Brooks, Dorothy Gheorghiu, Gordon He, Matthew Weinrub, Sze Ying Chan, Hye-Ri Kang, David Courtney, Jeffrey Everitt, Bryan R. Cullen, Dwight D. Koeberl
Abstract
Idiopathic pulmonary fibrosis (IPF) is a severe diffuse progressive fibrosing interstitial disease leading to respiratory failure and death in the absence of organ transplantation. Substantial evidence has confirmed the pivotal role of fibroblasts in the progression of IPF, yet effective therapeutic options are scarce. Single-cell transcriptomics profiling revealed that among the diverse fibroblast subsets, FAP1+ alveolar fibroblasts (AFs) are pivotal for the progression of IPF. On the basis of these findings, we developed FAP1-targeting chimeric antigen receptor cytotoxic effector regulatory T (CAR-cTregs) cells, which leverage the targeted killing advantage of the currently trending CAR-based immunotherapy for tumors and incorporate the immunosuppressive functions of Tregs to mitigate the inflammation caused by both the disease itself and CAR-T-cell infusion. Accordingly, CAR-cTregs were constructed to effectively eliminate FAP1+ fibroblasts in vitro. This cytotoxic effect can be abrogated by inhibitors of the granzyme-perforin pathway. In the bleomycin-induced PF model, CAR-cTregs were found to reverse fibrosis characterized by diminished recruitment of fibrocytes and improved remodeling of epithelial cells. Together, our results demonstrate that CAR-cTregs can serve as a promising therapeutic option for IPF and provide a novel strategy for treating multiple chronic inflammatory diseases by inducing both cytotoxicity and immunosuppression.
Authors
Yun-Han Jiang, Meng Zhou, Meng-Di Cheng, Sai Chen, Ying-Qiang Guo
Abstract
Androgen receptor positive prostate cancer (PC), castration resistant prostate cancer (CRPC) and neuroendocrine prostate cancer (NEPC) invariably become resistant to treatment with targeted and cytotoxic agents. Multiple pathways have been identified as being responsible for these pleotropic mechanisms of resistance. The MUC1 gene is aberrantly expressed in CRPC/NEPC in association with poor clinical outcomes; whereas, it is not known if the oncogenic MUC1-C/M1C protein drives treatment resistance. We demonstrated that MUC1-C is necessary for resistance of (i) PC cells to enzalutamide (ENZ), and (ii) CRPC and NEPC cells to docetaxel (DTX). Our results showed that MUC1-C-mediated resistance is conferred by upregulation of aerobic glycolysis and suppression of reactive oxygen species necessary for self-renewal. Dependence of these resistant phenotypes on MUC1-C for the cancer stem cell (CSC) state identified a potential target for treatment. In this regard, we further demonstrated that targeting MUC1-C with a M1C antibody-drug conjugate (ADC) is highly effective in suppressing (i) self-renewal of drug-resistant CRPC/NEPC CSCs and (ii) growth of t-NEPC tumor xenografts derived from drug-resistant cells and a patient with refractory disease. These findings uncovered a common MUC1-C-dependent pathway in treatment-resistant CRPC/NEPC progression and identified MUC1-C as a target for their treatment with a M1C ADC.
Authors
Keisuke Shigeta, Tatsuaki Daimon, Hiroshi Hongo, Sheng-Yu Ku, Hiroki Ozawa, Naoki Haratake, Atsushi Fushimi, Ayako Nakashoji, Atrayee Bhattacharya, Shinkichi Takamori, Michihisa Kono, Masahiro Rokugo, Yuto Baba, Takeo Kosaka, Mototsugu Oya, Justine Jacobi, Mark D. Long, Himisha Beltran, Donald W. Kufe
No posts were found with this tag.


