Research ArticleAgingMetabolism
Open Access | ![]() 10.1172/jci.insight.189683
10.1172/jci.insight.189683
Mutation in IR or IGF1R produces features of long-lived mice while maintaining metabolic health
Ulalume Hernández-Arciga,1 Jun Kyoung Kim,1,2 Jacob L. Fisher,1,2 Alexander Tyshkovskiy,3,4 Alibek Moldakozhayev,3,5,6,7 Catherine Hall,8 Souvik Ghosh,1,9 Yashvandhini Govindaraj,1,2 Ian J. Sipula,10,11 Jake Kastroll,10,11 Diana Cooke,12 Jinping Luo,13 Jonathan K. Alder,14,15 Stacey J. Sukoff Rizzo,1 Gene P. Ables,12 Eunhee Choi,8 Vadim N. Gladyshev,3,7 Michael J. Jurczak,10,11 Marc Tatar,16 and Andrey A. Parkhitko1,11
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Hernández-Arciga, U. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Kim, J. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Fisher, J. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Tyshkovskiy, A. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Moldakozhayev, A. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Hall, C. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Ghosh, S. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Govindaraj, Y. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Sipula, I. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Kastroll, J. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Cooke, D. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Luo, J. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by
Alder, J.
in:
PubMed
|
Google Scholar
|

1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Rizzo, S. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Ables, G. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Choi, E. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Gladyshev, V. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by
Jurczak, M.
in:
PubMed
|
Google Scholar
|
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Tatar, M. in: PubMed | Google Scholar
1Aging Institute of UPMC and University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
2Dietrich School of Arts & Sciences, University of Pittsburgh, Pennsylvania, USA.
3Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
4Belozersky Institute of Physico-Chemical Biology, Moscow State University, Moscow, Russia.
5Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada.
6Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada.
7Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
8Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, New York, USA.
9Department of Human Genetics, School of Public Health,
10Center for Metabolism and Mitochondrial Medicine, Department of Medicine, and
11Division of Endocrinology and Metabolism, Department of Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
12Orentreich Foundation for the Advancement of Science Inc., Cold Spring, New York, USA.
13Department of Molecular Biology, Cell Biology & Biochemistry, Brown University, Providence, Rhode Island, USA.
14Pulmonary, Allergy, Critical Care, and Sleep Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA.
15Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, USA.
16Department of Ecology, Evolution and Organismal Biology, and The Center on the Biology of Aging, Brown University, Providence, Rhode Island, USA.
Address correspondence to: Marc Tatar, Department of Ecology, Evolutionary Biology and Organismal Biology, Box G-W, Brown University, Providence, Rhode Island, 02912, USA. Phone: 401.863.3455; Email: marc_tatar@brown.edu. Or to: Andrey A Parkhitko, Aging Institute, Room 569, Bridgeside Point I, 100 Technology Dr., Pittsburgh, Pennsylvania, 15219, USA. Phone: 339.368.4594; Email: aparkhitko@pitt.edu.
Authorship note: MT and AAP contributed equally to this work.
Find articles by Parkhitko, A. in: PubMed | Google Scholar
Published November 11, 2025 - More info
JCI Insight. 2025;10(24):e189683. https://doi.org/10.1172/jci.insight.189683.
© 2025 Hernández-Arciga et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: December 3, 2024; Accepted: October 30, 2025
-
Results
Viable offspring with Arg>Cys substitutions in the KID of IR (InsRR1109C) or IGF1R (IGF-1RR1096C). The longevity inducing Arg>Cys substitution within the KID of dInr resides in the Arg-Pro-Glu sequence at the start of the KID (5). In general, KIDs are unstructured and variable domains that connect the N- and C-terminal lobes of receptor tyrosine kinases, including mammalian IR and IGF-1R (Figure 1A) (5, 25). We generated 2 knock-in mouse strains in the C57BL/6J background that carry the homologous KID Arg>Cys substitution in murine IR (InsRR1109C) or IGF-1R (IGF-1RR1096C) (Figure 1B). These mutations are viable as heterozygotes over a WT receptor allele. Parental crosses of WT with heterozygous InsRR1109C produced F1 progeny as 45.5% homozygote WT and 54.5% heterozygote. Crosses of WT with heterozygous IGF-1RR1096C adults produced 53.1% WT and 46.9% heterozygote offspring. Crosses of heterozygote InsRwt/InsRR1109C parents produced F1 in proportions of 27.7% WT, 63.8% heterozygote, and 8.5% mutant homozygote. Offspring of 38.9% WT, 52.8% heterozygote, and 8.3% mutant homozygotes were derived from crosses of heterozygote InsRwt/IGF-1RR1096C parents (Figure 1C). We could not generate any InsRR1109C, IGF-1RR1096C double heterozygous pups.
 or IGF1R (IGF-1RR1096C).") Figure 1
Figure 1Knock-in mouse strains carrying the homologous dInRKID Arg/Cys substitution in the KID of IR (InsRR1109C) or IGF1R (IGF-1RR1096C). (A) Sequence alignment of the KID domain for Drosophila, human, and mouse insulin receptor (IR) and insulin growth factor-1 receptor (IGF-1R). (B) DNA sequence chromatogram showing 1 peak for either AGG (WT) or TGC (homozygous) mutant and 2 peaks for heterozygous mutants. (C) Expected versus observed genotype ratios for offspring of WT × heterozygote and heterozygote × heterozygote.
Normal growth of InsRR1109C and IGF-1RR1096C heterozygotes. In Drosophila, growth is not retarded in WT/KID heterozygotes (dInR+/dInRKID) (5). We therefore assessed how growth through 4 months of age was affected when mice were heterozygous or homozygous for InsRR1109C or IGF-1RR1096C (Figure 2, A–D). Because we derived few F1 homozygotes, hereafter, we include those observations without statistical analyses. InsR+/InsRR1109C heterozygotes gained the same mass as WT siblings, while mutant homozygotes at first gained the same mass as WT but plateaued at a lighter weight (38% decrease in females, 48% decrease in males) (Figure 2E). Estimated growth rate based on a parametric model determined the rates were similar among WT and InsRR1109C heterozygotes (Figure 2, A, B, and E).
 Figure 2
Figure 2Effect of InsRR1109C and IGF-1RR1096C substitutions on growth rate. (A–D) Weekly mass (left panel) and logistic growth curves (right panel) in (A) InsRR1109C female, (B) InsRR1109C male, (C) IGF-1RR1096C female, and (D) IGF-1RR1096C male mice. (E and J) Female and male body weight in 4-month-old (E) InsRR1109C and (J) IGF-1RR1096C mice. (F and K) Female and male total lean mass in (F) InsRR1109C and (K) IGF-1RR1096C mice. (G and L) Female and male percent lean mass normalized to body weight in (G) InsRR1109C and (L) IGF-1RR1096C mice. (H and M) Female and male total fat mass weight in (H) InsRR1109C and (M) IGF-1RR1096C mice. (I and N) Female and male percent fat mass normalized to body weight in (I) InsRR1109C and (N) IGF-1RR1096C mice. WT and heterozygotes were available for statistical comparison (females WT/WT and WT/InsR n = 10; males WT/WT n = 10 and WT/InsR n = 12; females WT/WT n = 5 and WT/IGF-1R n = 6; males WT/WT n = 9 and WT/IGF-1R n = 10). Homozygote mutants were rare. Data are shown as mean ± SD, Student’s t test. *P < 0.05; **P < 0.01.
Female IGF-1RR1096C heterozygotes accumulated less mass than WT at 4 months (12.7% decrease); male heterozygotes weighed as much as WT (Figure 2, C, D, and J). The mass of IGF-1RR1096C homozygotes was reduced in both sexes (Figure 2, C, D, and J). The growth rate of IGF-1RR1096C heterozygotes equaled that of WT in males but was 0.63-fold reduced in females.
We used NMR to measure how the genotypes affected body composition. InsRR1109C and IGF-1RR1096C heterozygotes had the same percentage lean mass as WT (Figure 2, G and L). Total lean mass was slightly reduced in IGF-1RR1096C heterozygotes of both sexes (Figure 2, F and K). Fat mass (total and percentage) was moderately increased in male InsRR1109C heterozygotes (Figure 2, H and I), while total fat was reduced in female IGF-1RR1096C heterozygotes (Figure 2, M and N). Overall, heterozygotes with an Arg>Cys substitution in the KID of murine IR and IGF1R show few or modest effects on growth.
Normal metabolic rates in heterozygote InsRR1109C and IGF-1RR1096C. We used indirect calorimetry to evaluate how InsRR1109C and IGF-1RR1096C affect whole animal metabolism by measuring energy expenditure (EE), respiratory exchange ratio (RER), total activity, and food uptake in mice at 4 months of age. RER describes the substrates used to generate energy (26–30). The RER of WT heterozygotes with InsRR1109C or IGF-1RR1096C was like that of WT in both sexes (0.8–0.85), indicative of substrates from a mixture of protein, lipid, and carbohydrate (Figure 3, A and B, and Supplemental Figure 1, A–D; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.189683DS1). RER was reduced (<0.7) among the available male InsRR1109C homozygotes, indicating these animals used lipids rather than carbohydrates to produce energy as occurs when cells are insulin resistant (31) (Figure 3A and Supplemental Figure 1B). Resting EE (Figure 3, C and D, and Supplemental Figure 1, E–H) total activity (Figure 3, E and F, and Supplemental Figure 1, I–L), and food intake (Figure 3, G and H, and Supplemental Figure 1, M–P) did not differ among WT and InsRR1109C or IGF-1RR1096C heterozygote siblings. Overall, InsRR1109C and IGF-1RR1096C heterozygotes present metabolic profiles indistinguishable from their WT littermates.
 Figure 3
Figure 3Metabolic rate of InsRR1109C and IGF-1RR1096C heterozygous mice. (A and B) Respiratory exchange ratio (RER) in (A) InsRR1109C and (B) IGF-1RR1096C mice, 24-hour average (per kg of lean mass). (C and D) Energy expenditure (EE), 24 hr average (per kg of lean mass) in (C) InsRR1109C and (D) IGF-1RR1096C. (E and F) Total activity 24-hour average in (E) InsRR1109C and (F) IGF-1RR1096C. (G and H) Feeding, 24-hour average (per kg of lean mass) in (G) InsRR1109C and (H) IGF-1RR1096C. WT and heterozygous mice were available for statistical comparison (females WT/WT and WT/InsR n = 10; males WT/WT n = 10 and WT/InsR n = 12; females WT/WT n = 5 and WT/IGF-1R n = 6; males WT/WT n = 9 and WT/IGF-1R n = 10). Homozygous mutant mice were rare. Data are shown as mean ± SD, Student’s t test.
Normal glycated hemoglobin in heterozygote InsRR1109C and IGF-1RR1096C. To assess glucose homeostasis in InsRR1109C and IGF-1RR1096C heterozygotes, we measured glucose tolerance, plasma insulin, and glycated hemoglobin. Plasma glucose and glycated hemoglobin were normal in male and female InsRR1109C heterozygotes, although mice within the glucose tolerance test (GTT) had elevated fasting insulin (Figure 4, A–H, and Supplemental Figure 2, A–D). As expected, available homozygotes presented elevated plasma glucose, insulin, and glycated hemoglobin, consistent with a state of insulin resistance (Figure 4, A–H, and Supplemental Figure 2, A–D). Male and female IGF-1RR1096C heterozygotes presented normal glucose tolerance (GTT), glycated hemoglobin, and insulin titer except for fasted females where plasma insulin was reduced relative to WT (Figure 4, I–P, and Supplemental Figure 2, E–H).
 Figure 4
Figure 4Glycated hemoglobin in InsRR1109C and IGF-1RR1096C heterozygous mice. (A, E, I, and M) Blood glucose level measured during glucose tolerance test (GTT) using 1.5g/kg glucose dose in (A) female InsRR1109C, (E) male InsRR1109C, (I) female IGF-1RR1096C, and (M) male IGF-1RR1096C. (B, F, J, and N) Area under the curve (AUC) for glucose in (B) female InsRR1109C, (F) male InsRR1109C, (J) female IGF-1RR1096C, and (N) male IGF-1RR1096C. (C, G, K, and O) Fasted plasma insulin levels in (C) female InsRR1109C, (G) male InsRR1109C, (K) female IGF-1RR1096C, and (O) male IGF-1RR1096C. (D, H, L, and P) Glycated hemoglobin A1C percentage in (D) female InsRR1109C, (H) male InsRR1109C, (L) female IGF-1RR1096C, and (P) male IGF-1RR1096C. WT and heterozygous were available for statistical comparison (females WT/WT n = 9–10 and WT/InsR n = 9–11; males WT/WT n = 8–10 and WT/InsR n = 9–12; females WT/WT n = 7 and WT/IGF-1R n = 6; males WT/WT n = 9–10 and WT/IGF-1R n = 10–11). Homozygous mutant mice were rare. Data are shown as mean ± SD, Student’s t test. * P < 0.05; **P < 0.01; ***P < 0.001.
Overall, when heterozygous, the murine KID mutation in InsRR1109C results in moderate, compensatory hyperinsulinemia, while a heterozygote with the KID mutation IGF-1RR1096C has little effect on energy metabolism and glucose homeostasis.
Improved neuromuscular function in heterozygotes of InsRR1109C and IGF-1RR1096C. We assessed neuromuscular function of young mutant mice through rotarod agility, open field exploratory activity, and spontaneous wheel running. The ability to maintain balance on the accelerating rotarod did not differ between InsRR1109C or IGF-1RR1096C heterozygotes relative to their age- and sex-matched littermate WT controls (Figure 5, A and B). Motor coordination was intact in homozygous IGF-1R mice relative to heterozygotes and WT, although InsR homozygous appeared to be strongly impaired (based on our limited sample size) (Figure 5, A and B). In the open field test, which measures spontaneous exploratory activity, IGF-1RR1096C heterozygotes traveled the same distance as age- and sex-matched WT littermates. Interestingly, female InsRR1109C heterozygotes but not males traveled less than age- and sex-matched WT littermates (Figure 5, C and D). Rearing behavior, a typical exploratory behavior in mice that is measured by cumulative vertical activity in the open field, was increased in IGF-1RR1096C male heterozygotes but not female IGF-1RR1096C heterozygotes relative to age- and sex-matched WT littermate controls (Figure 5F). Cumulative vertical activity in the open field was unaltered in InsRR1109C heterozygotes relative to age- and sex-matched WT littermate controls (Figure 5E). Thigmotaxis behavior, measured by time spent at the perimeter of the open field, is an indicator of anxiety-like behavior and was similar across all sexes and genotypes (Figure 5, G and H). InsRR1109C and IGF-1RR1096C homozygotes had reduced cumulative distance traveled and rearing (based on limited observations) (Figure 5, C–F). Voluntary home cage wheel running assesses spontaneous physical activity. As expected, all patients were significantly more active during the dark cycle relative to the light cycle. InsRR1109C heterozygote males and females ran the same total distance. Male and female InsRR1109C heterozygotes were similar to WT with the exception of time spent running on the second night in males (Figure 5, I, J, M, and N). IGF-1RR1096C heterozygotes and WTs were similar in both total distance and time spent running (Figure 5, K, L, O, and P).
 Figure 5
Figure 5Neuromuscular function in InsRR1109C and IGF-1RR1096C heterozygous mice. (A and B) Latency to rotarod test fall in female and male (A) InsRR1109C and (B) IGF-1RR1096C. (C and D) Cumulative distance traveled in the open field test in female and male (C) InsRR1109C and (D) IGF-1RR1096C. (E and F) Cumulative Vertical Activity the open field test in female and male (E) InsRR1109C and (F) IGF-1RR1096C. (G and H) Time spent in margin areas in the open field test in female and male (G) InsRR1109C and (H) IGF-1RR1096C. (I, J, K, and L) Total distance traveled in wheel-running in (I) female InsRR1109C, (J) male InsRR1109C, (K) female IGF-1RR1096C, and (L) male IGF-1RR1096C. (M, N, O, and P) Total time spent wheel-running in (M) female InsRR1109C, (N) male InsRR1109C, (O) female IGF-1RR1096C, and (P) male IGF-1RR1096C. WT and heterozygous were available for statistical comparison (females WT/WT and WT/InsR n = 10; males WT/WT n = 10 and WT/InsR n = 10–12; females WT/WT n = 7 and WT/IGF-1R n = 6; males WT/WT n = 9 and WT/IGF-1R n = 9–10). Homozygous mutant mice were rare. Data are represented as means ± SD, Student’s t test. * P < 0.05.
Overall, young InsRR1109C heterozygotes perform slightly better as males in wheel running (males) but less so as females. IGF-1RR1096C heterozygote males have modestly elevated activity in open field as measured by rearing behavior.
Aging-associated hormones of InsRR1109C and IGF-1RR1096C heterozygotes. We measured plasma titers of FGF21, IGF-1, adiponectin, leptin, and GDF15 to assess how InsRR1109C and IGF-1RR1096C affect hormones that are sometimes associated with successful aging. We quantified plasma cholesterol and triglycerides to assess metabolic health. In all assays, plasma was collected from 4-month-old mice in the morning after 6 hours fasting.
FGF21 was increased in heterozygote InsRR1109C males but decreased in females, while FGF21 titer in IGF-1RR1096C heterozygous mice was equal to that of WT (Figure 6, A and B). Adiponectin was increased in both male and female InsRR1109C heterozygotes but did not differ between WT and heterozygotes of IGF-1RR1096C (Figure 6, C and D). IGF-1 levels were not different among InsRR1109C heterozygotes and WTs in either sex, but the hormone was significantly decreased in IGF-1RR1096C male heterozygotes (Figure 6, E and F). The satiety hormone leptin was elevated in male InsRR1109C heterozygotes, even though these mice have normal weight and food intake. In contrast, leptin was significantly decreased in IGF-1RR1096C female heterozygotes but not in males (Figure 6, G and H). The titer of GDF15 typically increases with chronological age and stress in WT mice (32–34). Here, the titer at 4 months was less in IGF-1RR1096C heterozygote females than in WT but not in males. GDF15 did not significantly differ between InsRR1109C heterozygotes and WT (Figure 6, I and J). Plasma triglycerides were significantly decreased in IGF-1RR1096C female heterozygotes but not in males or in InsRR1109C heterozygotes (Figure 6, K and L). We found no significant differences in plasma total cholesterol among genotype or sex (Figure 6, M and N).
 Figure 6
Figure 6Hormonal and metabolite profiles in InsRR1109C and IGF-1RR1096C heterozygous mice. (A–L) Hormone plasma levels in female and male mice from InsRR1109C and IGF-1RR1096C mice respectively, of FGF21 (A and B), Adiponectin (C and D), IGF-1 (E and F), Leptin (G and H), GDF15 (I and J), Triglycerides (TG) (K and L), and Total cholesterol (TC) (M and N). WT and heterozygous were available for statistical comparison (females WT/WT and WT/InsR n = 10; males WT/WT n = 9–10 and WT/InsR n = 11–12; females WT/WT n = 7 and WT/IGF-1R n = 5–6; males WT/WT n = 10 and WT/IGF-1R n = 9–10). Homozygous mutant mice were rare. Data are shown as mean ± SD, Student’s t test. *P < 0.05; **P < 0.01.
Overall, young InsRR1109C and IGF-1RR1096C heterozygous adults present some hormonal changes that are seen in established models of retarded murine aging (elevated FGF21, elevated adiponectin, decreased IGF-1). These patterns, however, are quite variable among genotypes and sex dependent, as seen for elevated FGF21 and leptin in InsRR1109C males as well as reduced GDF15 and plasma triglycerides in IGF-1RR1096C females.
InsRR1109C and IGF-1RR1096C heterodimer receptors have partial altered function. We determined how the KID mutation in IR or IGF-1R affects receptor function, measured as ligand-stimulated receptor kinase activity in human cell culture and by downstream signaling in murine tissue and in human cell culture.
We transfected IR/IGF-1R double-knockout 293FT cells with human IGF-1RWT alone, IGF-1RR1095C alone (corresponding to mouse IGF-1R R1096C, hereafter referred to as R1096C), or a 1:1 mixture of both plasmids to produce heterodimer receptors (Figure 7, A and B). To assess relative expression of these receptor types, we cotransfected Myc-tagged R1096C with untagged IGF-1RWT. IGF-1 stimulation robustly induced autophosphorylation of IGF-1RWT and phosphorylation of AKT and ERK. In contrast, R1096C homodimers exhibited markedly reduced autophosphorylation. Despite this defect, IGF-1 ligand still induced AKT phosphorylation to approximately 80% of the level observed in WT-expressing cells, whereas there is only weak phosphorylation of ERK (Figure 7, A and B). Assuming equal expression, cotransfection of IGF-1RWT and R1096C is expected to generate 25% WT:WT homodimers, 50% WT:R1096C heterodimers, and 25% R1096C:R1096C homodimers. If the heterodimer is inactive, IGF-1–induced autophosphorylation should resemble that of cells expressing R1096C alone. However, cells coexpressing WT and R1096C showed IGF-1R autophosphorylation at ~50% of WT levels and pERK activation at ~70% of those in WT-expressing cells, suggesting that the WT:R1096C heterodimer produces partial functional activity (Figure 7, A and B).
 Figure 7
Figure 7Receptor function in transfected 293FT cells with IGF-1R WT:R1096C heterodimers and IR WT:R1109C heterodimers. (A) IGF-1R signaling in response to the indicated concentrations of IGF-1 for 10 minutes in IR/IGF-1R knockout 293FT cells expressing IGF-1R WT and mutant receptors. (B) Quantitative analysis of the Western blot data. Levels of IGF-1R autophosphorylation were normalized to total IGF-1R levels and presented as intensities relative to those in IGF-1R WT cells treated with 50 nM IGF1. Data are shown as mean ± SD. Significance evaluated by 2-way ANOVA with interaction among n = 4 independent experiments, ***P < 0.0001. (C) IR signaling in response to concentrations of insulin for 10 minutes in IR/IGF-1R knockout 293FT cells expressing IR WT and mutant receptors. (D) Quantitative analysis of the Western blot data. Levels of IR autophosphorylation were normalized to total IR levels and presented as intensities relative to those in IR WT cells treated with 50 nM insulin. Data are shown as mean ± SD. Significance evaluated by 2-way ANOVA with interaction among n = 3 independent experiments, **P < 0.01; ***P < 0.001; ****P < 0.0001.
We performed similar experiments using human IRWT and an IRR1107C mutant (corresponding to mouse IR-A R1109C, hereafter referred to as R1109C) (Figure 7, C and D). R1109C homodimers showed greatly reduced autophosphorylation and impaired pERK, while pAKT remained comparable with IRWT homodimer cells. Coexpression of IRWT significantly restored autophosphorylation and pERK signaling in the presence of R1109C. Together, these results support the conclusion that both IGF-1R WT:R1096C and IR WT:R1109C heterodimers are functional whereby they retain kinase activity and partial ability to engage downstream signaling pathways.
We likewise assessed signal transduction pathways (pERK/ERK and pAKT/AKT) from liver tissue of InsRR1109C and IGF-1RR1096C heterozygote mice. We also measured pACC/ACC and pS6/S6 to describe altered AMPK and mTORC1 activity, which are characteristic of some long-lived mice (35–39).
pErk/Erk levels were significantly higher in livers of male and female InsRR1109C heterozygotes (Supplemental Figure 3, A–D) but were not affected in IGF-1RR1096C heterozygotes (Supplemental Figure 3, A–D). pACC/ACC levels were normal in InsRR1109C females but significantly decreased in livers of male InsRR1109C heterozygotes (Supplemental Figure 3, A–D). In contrast, pACC/ACC was elevated in male and female IGF-1RR1096C heterozygotes (Supplemental Figure 3, A–D). pAKT/AKT was normal in InsRR1109C females and males (Supplemental Figure 3, A–D) but significantly decreased in IGF-1RR1096C females and increased in males (Supplemental Figure 3, A–D); pS6/S6 was significantly lower in InsRR1109C females but normal in InsRR1109C males (Supplemental Figure 3, A–D). pS6/S6 was reduced in female IGF-1RR1096C heterozygotes but was upregulated in male IGF-1RR1096C heterozygotes (Supplemental Figure 3, A–D).
Overall, the activity of TOR, AMPK, and MAPK signaling in mouse tissue was partially altered by InsRR1109C and IGF-1RR1096C when these mutants complement a WT receptor allele. Some of these changes (Table 1) are seen in other models of extended murine longevity, while other observations here are ambiguous, such as where male IGF-1RR1096C increase both AMPK activity and TOR activity.
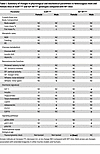 Table 1
Table 1Summary of changes in physiological and biochemical parameters in heterozygous male and female mice of InsRR1109C and IGF-1RR1096C genotypes compared with WT mice
Decreased biological age in livers of heterozygous female IGF-1RR1096C mice. In this preliminary report, we cannot assess aging through survival analysis. Accordingly, we use transcriptomic biomarkers as proxies of aging and mortality. Recently developed transcriptome-based clocks can predict age-associated mortality and identify the molecular pathways that mediate differences in biological age (40).
We focused on IGF-1R mice using RNA-Seq from gastrocnemius muscle and liver of WT and heterozygote IGF-1RR1096C of both sexes at 4 months of age. We did not include heterozygote InsRR1109C mice in the analysis because they develop mild hyperinsulinemia. PCA of muscle samples separated the samples based on sex but not by genotype (Supplemental Figure 4A). PCA of the liver tissue separated samples by sex on the first principal component and by genotype among females on the second principal axis (Figure 8A). This suggests there is a sex-specific effect of the IGF-1RR1096C KID within the liver. Accordingly, over 5,000 differentially expressed genes (Padj < 0.05) were identified in livers of IGF-1RR1096C heterozygous females, while few DEGs were seen in female muscle or in either tissue of males (Figure 8B, Supplemental Figure 4B, and Supplemental Table 1). Interestingly, the expression of Igf1 and Igf1r were significantly upregulated in the liver of female IGF-1RR1096C mice, while IGF-1 in plasma was significantly decreased in IGF-1RR1096C male heterozygotes (Supplemental Figure 4C).
 Figure 8
Figure 8Transcriptome-estimated biological age in liver of female IGF-1RR1096C heterozygote. (A) Principal component analysis (PCA) of liver gene expression from WT and IGF-1RR1096C heterozygous mice. (B) Volcano plots of gene expression changes induced in the livers of IGF-1RR1096C heterozygous female mice compared with WT. Benjamini-Hochberg (BH) Padj value threshold at 0.05 (dotted line). (C) Gene set enrichment analysis (GSEA) of transcriptomic changes induced in IGF-1RR1096C heterozygous mice relative to WT (blue), signatures of aging and mortality (red), and biomarkers of lifespan-extending interventions (green). Gene sets derived from KEGG, REACTOME, and HALLMARKS ontologies (full data in Supplemental Table 2). NES: normalized enrichment score. (D) Mortality transcriptomic age (tAge) of WT and IGF-1RR1096C heterozygous female mice pooled across skeletal muscle and liver, as assessed with the rodent multi-tissue Elastic Net (EN) clock. tAges were adjusted for tissue type using an ANOVA model, and the resulting residuals are shown. Group differences were assessed using ANOVA, with BH Padj values. (E) Standardized change in mortality tAge in IGF-1RR1096C heterozygous mice relative to sex-matched WT controls, assessed using module-specific transcriptomic clocks of expected mortality. OxPhos, Oxidative Phosphorylation; TCA, Tricarboxylic Acid; ER, Endoplasmic reticulum; UPR, Unfolded Protein Response; met, metabolism; ECM, Extracellular Matrix; EMT, Epithelial-mesenchymal transition. ^Padj < 0.1, *Padj < 0.05, **Padj < 0.01, ***Padj < 0.001.
We compared the transcriptional profiles from the female IGF-1RR1096C liver to characteristic signatures seen in established murine models of delayed aging (41, 42). This revealed a positive correlation with longevity-associated signatures and a negative correlation with aging- and mortality-associated biomarkers. We observed a positive correlation with the signature specific to calorie restriction (Padj < 0.001) and a negative association with signatures characteristic of aging-associated degeneration (kidney aging and rodent aging, each Padj < 0.001) (Supplemental Figure 4D). Gene set enrichment analysis (GSEA) from IGF-1RR1096C liver samples revealed a negative normalized enrichment score (NES) for metabolic processes (fatty acid metabolism, oxidative phosphorylation), complement, mTORC1 signaling, and hypoxia (Figure 8C and Supplemental Table 2). The observed downregulation of mTORC1-associated genes is consistent with our Western blot data where pS6 is decreased in IGF-1RR1096C livers (Supplemental Figure 3C).
We then applied multitissue transcriptomic clocks of expected mortality (40) to estimate transcriptomic age (tAge) in tissue samples of female WT and IGF-1RR1096C heterozygous animals. While individual organs did not show statistically significant differences, likely due to low sample size (Supplemental Figure 4E), female IGF-1RR1096C heterozygotes displayed significantly lower tAge when liver and muscle tissues were pooled (Figure 8D). We applied module-specific transcriptomic mortality clocks to gain mechanistic insight into the biological pathways contributing to this effect (40). Several transcriptomic modules showed reduced tAge in liver tissue of IGF-1RR1096C females (Padj < 0.05) (Figure 8D), but no significant differences were observed in other tissues or in males. Strongly affected modules included lipid metabolism, VEGF signaling, mRNA splicing, mitochondria, NRF2 signaling, adaptive immunity, amino acid metabolism, heat-stress response, and translation (Figure 8E). Modules related to IFN signaling and chromatin modification showed modest increases in tAge, suggesting that while antiaging effects are predominant in this model, some proaging signals may be present.
In summary, female IGF-1RR1096C heterozygous mice exhibit decreased biological age, particularly in the liver, highlighting potential molecular pathways contributing to the health-promoting effect of this genetic model, and this finding warrants an actual lifespan experiment.
 or IGF1R (IGF-1RR1096C).")










