Research ArticleMicrobiologyOncology
Open Access | ![]() 10.1172/jci.insight.176839
10.1172/jci.insight.176839
Characterization of the vaginal microbiome of postmenopausal patients receiving chemoradiation for locally advanced cervical cancer
Brett A. Tortelli,1,2 Jessika Contreras,1 Stephanie Markovina,1,3 Li Ding,3,4,5,6 Kristine M. Wylie,7 and Julie K. Schwarz1,3,8
1Department of Radiation Oncology, Washington University School of Medicine, St. Louis, Missouri, USA.
2Transitional Year Program, Mercy Hospital St. Louis, St. Louis, Missouri, USA.
3Alvin J. Siteman Cancer Center,
4Department of Medicine,
5McDonnell Genome Institute,
6Department of Genetics,
7Department of Pediatrics, and
8Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Julie K. Schwarz, Department of Radiation Oncology, Washington University School of Medicine, 4511 Forest Park Avenue, St. Louis, Missouri, 63108, USA. Phone: 314.273.0275; Email: jschwarz@wustl.edu.
Find articles by Tortelli, B. in: PubMed | Google Scholar
1Department of Radiation Oncology, Washington University School of Medicine, St. Louis, Missouri, USA.
2Transitional Year Program, Mercy Hospital St. Louis, St. Louis, Missouri, USA.
3Alvin J. Siteman Cancer Center,
4Department of Medicine,
5McDonnell Genome Institute,
6Department of Genetics,
7Department of Pediatrics, and
8Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Julie K. Schwarz, Department of Radiation Oncology, Washington University School of Medicine, 4511 Forest Park Avenue, St. Louis, Missouri, 63108, USA. Phone: 314.273.0275; Email: jschwarz@wustl.edu.
Find articles by Contreras, J. in: PubMed | Google Scholar
1Department of Radiation Oncology, Washington University School of Medicine, St. Louis, Missouri, USA.
2Transitional Year Program, Mercy Hospital St. Louis, St. Louis, Missouri, USA.
3Alvin J. Siteman Cancer Center,
4Department of Medicine,
5McDonnell Genome Institute,
6Department of Genetics,
7Department of Pediatrics, and
8Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Julie K. Schwarz, Department of Radiation Oncology, Washington University School of Medicine, 4511 Forest Park Avenue, St. Louis, Missouri, 63108, USA. Phone: 314.273.0275; Email: jschwarz@wustl.edu.
Find articles by
Markovina, S.
in:
PubMed
|
Google Scholar
|

1Department of Radiation Oncology, Washington University School of Medicine, St. Louis, Missouri, USA.
2Transitional Year Program, Mercy Hospital St. Louis, St. Louis, Missouri, USA.
3Alvin J. Siteman Cancer Center,
4Department of Medicine,
5McDonnell Genome Institute,
6Department of Genetics,
7Department of Pediatrics, and
8Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Julie K. Schwarz, Department of Radiation Oncology, Washington University School of Medicine, 4511 Forest Park Avenue, St. Louis, Missouri, 63108, USA. Phone: 314.273.0275; Email: jschwarz@wustl.edu.
Find articles by Ding, L. in: PubMed | Google Scholar
1Department of Radiation Oncology, Washington University School of Medicine, St. Louis, Missouri, USA.
2Transitional Year Program, Mercy Hospital St. Louis, St. Louis, Missouri, USA.
3Alvin J. Siteman Cancer Center,
4Department of Medicine,
5McDonnell Genome Institute,
6Department of Genetics,
7Department of Pediatrics, and
8Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Julie K. Schwarz, Department of Radiation Oncology, Washington University School of Medicine, 4511 Forest Park Avenue, St. Louis, Missouri, 63108, USA. Phone: 314.273.0275; Email: jschwarz@wustl.edu.
Find articles by Wylie, K. in: PubMed | Google Scholar
1Department of Radiation Oncology, Washington University School of Medicine, St. Louis, Missouri, USA.
2Transitional Year Program, Mercy Hospital St. Louis, St. Louis, Missouri, USA.
3Alvin J. Siteman Cancer Center,
4Department of Medicine,
5McDonnell Genome Institute,
6Department of Genetics,
7Department of Pediatrics, and
8Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, Missouri, USA.
Address correspondence to: Julie K. Schwarz, Department of Radiation Oncology, Washington University School of Medicine, 4511 Forest Park Avenue, St. Louis, Missouri, 63108, USA. Phone: 314.273.0275; Email: jschwarz@wustl.edu.
Find articles by
Schwarz, J.
in:
PubMed
|
Google Scholar
|
Published February 4, 2025 - More info
JCI Insight. 2025;10(6):e176839. https://doi.org/10.1172/jci.insight.176839.
© 2025 Tortelli et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: November 1, 2023; Accepted: January 31, 2025
-
Results
Study population. Twenty-six women who are postmenopausal receiving intracavitary brachytherapy as part of the standard-of-care chemoradiation for locally advanced (Stage IB2 or greater) cervical cancer were included in our analysis. The average age of women in this study was 63 years, with a range of 52–77 years. The patient cohort was predominantly White (80.8%), with fewer Black (11.5%) and Asian (7.5%) patients. Most (84.6%) were diagnosed with squamous cell carcinoma, and most (84.6%) were positive for high-risk HPV. Initial-staging PET imaging showed 6 patients (23.1%) had positive pelvic lymph nodes and 9 (34.6%) had positive aortic lymph nodes. All but 1 patient was treated with cisplatin chemotherapy. Demographic and clinical characteristics of the patient cohort are presented in Table 1.
Microbiome characterization. To characterize the composition of the microbiome, we performed 16S rRNA gene sequencing on 56 vaginal swab specimens collected from our cohort of patients. Of the 56 specimens, 25 were collected prior to chemoradiation (T1), and 31 were collected after chemoradiation was initiated; either 1–2 weeks after treatment start (T2) or 3 weeks after treatment start (T3). All but 5 T1 specimens and one T3 specimen had matched samples available from the same patient for comparison. We observed diverse communities with a median of 32 speciestaxa (interquartile range [IQR], 26–39) represented in each sample. Relative abundance data for each sample is presented in Supplemental Table 1 (supplemental material available online with this article; https://doi.org/10.1172/jci.insight.176839DS1). We generated stacked bar plots for each sample to illustrate the taxonomic composition of each community (Figure 1). Lactobacillus was uncommon, while anaerobes such as Prevotella, Porphyromonas, Peptoniphilus, Anaerococcus, and Fusobacterium were most abundant.
 Figure 1
Figure 116S rRNA gene community profiles. Stacked bar graph showing the proportion of each taxa in a sample. Only the top 21 taxa identified within all samples are represented; all other taxa are grouped as “Others” for ease of visualization. Sampling time point (T1 = pretreatment, T2 = 1–2 weeks into treatment, and T3 = 3 weeks into treatment) is designated along the x axis and grouped by patient.
Microbiome before and during chemoradiation. We compared the microbiome before and during chemoradiation therapy. To assess whether chemoradiation reduced the bacterial biomass we utilized a quantitative PCR (qPCR) assay to approximate the total bacterial abundance for each sample. In a paired analysis, we did not observe a significant difference in bacterial abundance before and during chemoradiation (P = 0.73) (Figure 2A). Among all samples, α diversity did not significantly differ by collection time point (P = 0.78). A paired analysis of T1 and T3 specimens also failed to show a difference in α diversity (P = 0.71) (Figure 2B).
 Figure 2
Figure 2Bacterial abundance and α diversity by sampling time point. (A and B) A paired analysis of samples taken pretreatment (T1) and 3 weeks into treatment (T3) for bacterial abundance (A) and α diversity (B). P values were calculated with a Wilcoxon signed-rank test.
Nonmetric dimensional scaling (NMDS) was used to plot and compare the similarity of the microbiome across individuals and sampling time points. Analysis of the NMDS data do not reveal any significant association between the taxonomic composition of the microbiome and sampling time point (P = 0.56). We observed that pretreatment and during-treatment samples collected from the same woman were more similar to one another than to samples collected from others at the same time point (Figure 3). To determine whether particular taxa were enriched or depleted by chemoradiation, we performed linear discriminant analysis (LDA) using patient microbiomes for which both T1 and T3 samples were available. However, we did not identify any notable enrichment or depletion of the relative abundance of taxa within this dataset. Analysis of compositions of microbiomes with bias correction (ANCOM-BC) is an alternative method for differential abundance analysis that corrects for sampling biases (16). We applied ANCOM-BC to the same dataset and confirmed the results obtained using LDA.
 Figure 3
Figure 3NMDS by time point. NMDS plot of taxonomic composition by sampling time point. Comparing samples taken prior to treatment (green), 1–2 weeks into treatment (orange), and 3 weeks into treatment (purple). Normal confidence ellipses represent 50% confidence level and centroid of data. Permutational multivariate ANOVA of distance matrices did not identify significant differences by sampling time point (P = 0.56).
Community characteristics by disease recurrence. We sought to identify whether certain characteristics of the pretreatment microbiome were correlated with treatment response as determined by disease recurrence during the follow-up period. Pretreatment microbiome characterization and recurrence data were available for 24 of the patients. Eleven (46%) of the patients experienced a recurrence during the follow-up period. The median follow-up time for patients alive at time of last follow-up was 50 months (range, 20–61 months). We did not observe any association between demographic or clinical characteristics and disease recurrence. A summary of demographic and clinical characteristics by recurrence status can be found in Table 2.
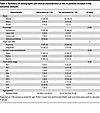 Table 2
Table 2Summary of demographic and clinical characteristics of the 24 patients included in the outcomes analysis.
We compared the pretreatment microbiomes of patients who developed disease recurrence (treatment nonresponders) to those who did not (treatment responders). There was no significant difference in bacterial abundance (P = 0.22) or community diversity (P = 0.58) between treatment responders and nonresponders (Figure 4). NMDS was used to plot and compare the microbiomes of treatment responders and nonresponders. Samples did not appear to cluster by recurrence status, and no significant difference was observed between treatment responders and nonresponders (P = 0.47) (Figure 5). In an effort to identify taxonomic biomarkers of treatment response within the microbiome, we correlated the relative abundance of the top 21 taxa with recurrence. Though it did not reach statistical significance, a greater relative abundance of Fusobacterium was observed among treatment nonresponders (median, 0.017; IQR, 0.002–0.041) than treatment responders (median, 0.001; IQR, 0.000–0.005) (unadjusted P = 0.046) (Figure 6). No other genera were identified to be enriched by treatment response (Supplemental Table 2). Linear discriminate analysis and ANCOM-BC corroborated these findings.
 Figure 4
Figure 4Bacterial abundance and α diversity by recurrence status. (A and B) An analysis of bacterial abundance (A) and α diversity (B) of pretreatment samples of patients who had disease recurrence and those that did not. P values were calculated with a Kruskal-Wallis rank sum test.
 Figure 5
Figure 5NMDS by recurrence status. NMDS plot of pretreatment taxonomic composition by recurrence status. Comparing samples from patients who had a recurrence (orange) and those who did not (green). Normal confidence ellipses represent 50% confidence level and centroid of data. Permutational multivariate ANOVA of distance matrices did not identify significant differences by sampling time point (P = 0.47).
 Figure 6
Figure 6Fusobacterium abundance by recurrence status. The relative abundance of Fusobacterium in pretreatment samples of patients who had disease recurrence and those who did not. P values were calculated with a Kruskal-Wallis rank sum test.










