Research ArticleAgingNeuroscience
Open Access | ![]() 10.1172/jci.insight.175917
10.1172/jci.insight.175917
Air pollution and Alzheimer disease phenotype deplete esterified proresolving lipid mediator reserves in the brain
Ameer Y. Taha,1,2,3 Qing Shen,1 Yurika Otoki,1,4 Nuanyi Liang,1 Kelley T. Patten,5 Anthony E. Valenzuela,5 Christopher D. Wallis,6 Douglas J. Rowland,7 Abhijit J. Chaudhari,7,8 Keith J. Bein,9,10 Anthony S. Wexler,6,10 Lee-Way Jin,11 Brittany N. Dugger,11 Danielle J. Harvey,12 and Pamela J. Lein5,13
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Taha, A. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Shen, Q. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Otoki, Y. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Liang, N. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Patten, K. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Valenzuela, A. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Wallis, C. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Rowland, D. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by
Chaudhari, A.
in:
PubMed
|
Google Scholar
|

1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Bein, K. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Wexler, A. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Jin, L. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Dugger, B. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Harvey, D. in: PubMed | Google Scholar
1Department of Food Science and Technology, College of Agriculture and Environmental Sciences,
2Center for Neuroscience, and
3West Coast Metabolomics Center, Genome Center, UCD, Davis, California, USA.
4Food and Biodynamic Laboratory, Graduate School of Agricultural Science, Tohoku University, Sendai, Miyagi, Japan.
5Department of Molecular Biosciences, School of Veterinary Medicine,
6Air Quality Research Center,
7Center for Genomic and Molecular Imaging, UCD, Davis, California, USA.
8Department of Radiology, School of Medicine, UCD, Sacramento, California, USA.
9Center for Health and the Environment and
10Departments of Mechanical and Aerospace Engineering, Civil and Environmental Engineering, and Land, Air and Water Resources, UCD, Davis, California, USA.
11Department of Pathology and Laboratory Medicine, UCD School of Medicine, Sacramento, California, USA.
12Department of Public Health Sciences, UCD, Davis, California, USA.
13The MIND Institute, School of Medicine, UCD, Sacramento, California, USA.
Address correspondence to: Ameer Y. Taha, Department of Food Science and Technology, College of Agriculture and Environmental Sciences, 1 Shields Ave. University of California, Davis, California, 95616, USA. Phone: 530.752.7096; Email: ataha@ucdavis.edu.
Find articles by Lein, P. in: PubMed | Google Scholar
Published May 13, 2025 - More info
JCI Insight. 2025;10(15):e175917. https://doi.org/10.1172/jci.insight.175917.
© 2025 Taha et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: December 26, 2023; Accepted: May 7, 2025
-
Results
Effects of AD genotype and TRAP exposure on NL-bound oxylipins in brain of 15-month-old rats. In order to assess the effects of AD genotype or TRAP exposure on NL-bound oxylipins, rats expressing mutations in APPswe and PS1ΔE9, henceforth called AD transgenic rats or WT rats, were exposed to FA or TRAP from 1 to 15 months of age as shown in Figure 1. Esterified oxylipins were captured using the mass spectrometry (MS) metrics shown in Supplemental Table 1 and imputation parameters for missing values shown in Supplemental Table 2.
 Figure 1
Figure 1Overall study design. Male and female rats were exposed to filtered air (FA) or traffic-related air pollution (TRAP) from 1 to 15 months of age. After euthanasia, the brains were subjected to lipidomic analysis of neutral lipid (NL) and phospholipid (PL) bound oxylipins.
AD transgenic rats showed significant changes in NL-bound oxylipins in females but not males (P < 0.05 for main effects of sex and genotype; Supplemental Table 3). As shown in Figure 2, compared with WT female rats, significant reductions in NL-bound oxylipin were observed in TgF344-AD female rats exposed to either FA or TRAP (P < 0.05 by 1-way ANOVA), whereas only a few significant changes were detected in males (Supplemental Table 4; P < 0.05 by 1-way ANOVA).
 female rats exposed to FA or TRAP for 14 months.") Figure 2
Figure 2Oxylipin concentrations in brain NL of 15-month-old WT or TgF344-AD (Tg) female rats exposed to FA or TRAP for 14 months. Data are shown as mean ± SD of n = 7 WT-FA, n = 7 Tg-FA, n = 6 WT-TRAP, and n = 7 Tg-TRAP. Data were analyzed by 1-way ANOVA followed by Duncan’s post hoc test. Different-letter superscripts indicate that the means differed significantly from each other at P < 0.05. (A–E) DGLA-derived oxylipins (A), ALA-derived oxylipins (B), EPA-derived oxylipins (C), AA-derived oxylipins (D), and DHA-derived oxylipins (E). DiHETE, dihydroxyeicosatetraenoic acid; DiHETrE, dihydroxyeicosatrienoic acid; DiHOME, dihydroxyoctadecenoic acid; DiHDPA, dihydroxydocosapentaenoic acid; EpDPE, epoxydocosapentaenoic acid; EpETE, epoxyeicosatetraenoic acid; EpETrE, epoxyeicosatrienoic acid; EpOME, epoxyoctadecenoic acid; HDoHE, hydroxydocosahexaenoic acid; HEPE, hydroxyeicosapentaenoic acid; HETE, hydroxyeicosatetraenoic acid; HETrE, hydroxyeicosatrienoic acid; HODE, hydroxyoctadecadienoic acid; HOTrE, hydroxyoctadecatrienoic acid; oxo-ETE, oxo-eicosatetraenoic acid; oxo-ODE, oxo-octadecadienoic acid; TriHOME, trihydroxyoctadecenoic acid.
Most of the changes in females were observed in oxylipins involved in inflammation resolution (e.g., EpETrEs, EpETEs, and EpDPEs) or oxylipins destined toward proresolving lipid mediator synthesis (e.g., HETE precursors to lipoxins). As shown in Figure 2, dihomo-γ-linoleic acid–derived (DGLA-derived) 15(S)-HETrE (Figure 2A); EPA-derived 17(18)-EpETE and 11(12)-EpETE (Figure 2C); AA-derived 20-HETE, 15-HETE, 11-HETE, 11(12)-EpETrE, and 14,15-DiHETrE (Figure 2D); and DHA-derived 19(20)-EpDPE, 16(17)-EpDPE, 13(14)-EpDPE, 10(11)-EpDPE, 7(8)-EpDPE, and 19,20-DiHDPA (Figure 2E) were significantly lower by 22%–43% in Tg-FA rats compared with WT-FA controls (P < 0.05). The majority of these oxylipins [AA-derived 11(12)-EpETrE and 14,15-DiHETrE, and DHA-derived 19(20)-EpDPE, 16(17)-EpDPE, 13(14)-EpDPE, 7(8)-EpDPE, and 19,20-DiHDPA], as well as AA-derived 5-oxo-ETE and 11,12-DiHETrE, and DHA-derived 16,17-DiHDPA, were also lower by 8%–43% in Tg-TRAP exposed rats compared with WT-FA or WT-TRAP, suggesting an AD-effect, independent of TRAP exposure. α-Linolenic acid–derived (ALA-derived) 13-hydroxyoctadecatrienoic acid (13-HOTrE) was 4-fold higher in Tg-TRAP compared with WT-TRAP rats (Figure 2B; P < 0.05), but neither groups differed significantly from WT-FA controls.
TRAP exposure alone minimally affected NL oxylipins in WT rats. The few observed changes included a significant increase in AA-derived 12-oxo-ETE by ~2-fold in WT-TRAP rats compared with WT-FA, Tg-FA, and Tg-TRAP rats (Figure 2D), and there was a significant 22% decrease in DHA-derived 7(8)-EpDPE in WT-TRAP rats compared with WT-FA controls (Figure 2E).
Overall, the data suggest that the AD genotype reduced multiple proresolving oxylipins in NL of female rats. Of the ~17 significantly altered oxylipins in TgF344-AD rats exposed to FA or TRAP, 11 (65%) have proresolving effects in vivo, 2 (11-HETE, 20-HETE) have proinflammatory effects, and 4 are sEH inactivation products of fatty acid epoxides (11,12-DiHETrE, 14,15-DiHETrE, 16,17-DiHDPA, and 19,20-DiHDPA).
Effects of AD genotype and TRAP exposure on PL-bound oxylipins in brain of 15-month-old rats. We next examined the effects of AD or TRAP exposure on PL-bound oxylipins. There were no significant effects of AD genotype or TRAP exposure in males (Supplemental Table 5). However, significant changes due to genotype or TRAP were observed in females mainly (Supplemental Table 6) as depicted in Figure 3.
 female rats exposed to FA or TRAP for 14 months.") Figure 3
Figure 3Oxylipin concentrations in brain PL of 15-month-old WT or TgF344-AD (Tg) female rats exposed to FA or TRAP for 14 months. Data are shown as mean ± SD of n = 7 WT-FA, n = 7 Tg-FA, n = 6 WT-TRAP, and n = 7 Tg-TRAP. Data were analyzed by 1-way ANOVA followed by Duncan’s post hoc test. Different-letter superscripts indicate that the means differed significantly from each other at P < 0.05. (A–D) LA-derived oxylipins (A), AA-derived oxylipins (B), EPA-dedrived oxylipins (C), and DHA-derived oxylipins (D). DiHETE, dihydroxyeicosatetraenoic acid; DiHETrE, dihydroxyeicosatrienoic acid; DiHOME, dihydroxyoctadecenoic acid; DiHDPA, dihydroxydocosapentaenoic acid; EpDPE, epoxydocosapentaenoic acid; EpETE, epoxyeicosatetraenoic acid; EpETrE, epoxyeicosatrienoic acid; EpOME, epoxyoctadecenoic acid; HDoHE, hydroxydocosahexaenoic acid; HEPE, hydroxyeicosapentaenoic acid; HETE, hydroxyeicosatetraenoic acid; HETrE, hydroxyeicosatrienoic acid; HODE, hydroxyoctadecadienoic acid; HOTrE, hydroxyoctadecatrienoic acid; oxo-ETE, oxo-eicosatetraenoic acid; oxo-ODE, oxo-octadecadienoic acid; TriHOME, trihydroxyoctadecenoic acid; PG, prostaglandin.
AD genotype significantly reduced linoleic acid– (LA-), AA-, EPA-, and DHA-derived oxylipins. Compared with WT-FA controls, the Tg-FA group exhibited a significant reduction in LA-derived 13-oxo-ODE, 9-oxo-ODE, and 9(10)-EpOME (by 31%–40%; Figure 3A); AA-derived 5-,8-.9-,11-,12- and 15-HETEs; 12- and 15-oxo-ETE; 8(9)-EpETrE; 5,6-DiHETrEs, 8,9-DiHETrEs, 11,12-DiHETrEs, and 14,15-DiHETrEs; PGE2, and PGB2 (by 27%–63%; Figure 3B); EPA-derived 15-HEPE (by ~42%; Figure 3C); and DHA-derived 17-hydroxydocosahexaenoic acid (17-HDoHE), 19(20)-EpDPE, 19,20-DiHDPA, and 16,17-DiHDPA (by 24%–42%; Figure 3D). Similar significant reductions in PL-bound oxylipins were observed in Tg-TRAP rats compared with WT-FA controls.
TRAP exposure alone resulted in similar reductions in AA-, EPA-, and DHA-derived PL-bound oxylipins in WT rats. Compared with WT-FA controls, WT-TRAP rats showed significant reductions in AA-derived 15-HETE, 11-HETE, 9-HETE, 5-HETE, 14,15-DiHETrE, 11,12-DiHETrE, and 5,6-DiHETrE by 23%–40% (Figure 3B; P < 0.05); EPA-derived 15-HEPE by 27% (Figure 3C; P < 0.05); and DHA-derived 17-HDoHE, 19,20-DiHDPA, and 16,17-DiHDPA by 29% to 37 % (Figure 3D; P < 0.05).
Of the 23 significantly altered oxylipin in PL by AD genotype and/or TRAP exposure, 7 (30%) are proresolving or serve as precursors to proresolving mediators [5-HETE, 15-HETE, 12-oxo-ETE, 15-oxo-ETE, 8(9)-EpETrE, 15-HEPE, 17-HDoHE], 8 are proinflammatory, 6 are diols of sEH epoxide metabolism, and 2 are prostanoids.
Association between rat brain in vivo imaging measures of AD markers of pathophysiology and esterified oxylipins. We next explored the relationship between brain esterified oxylipins and markers of AD neuropathology (TSPO, SV2A, and fluorodeoxyglucose) in females. There were no significant group differences between the groups in MRI measures of brain atrophy or PET uptake values for tracers targeted toward TSPO (marker of neuroinflammation), SV2A (marker of synaptic density), and fluorodeoxyglucose (marker of glucose metabolism) in 15-month-old female rats (Table 1). PL-bound 17(18)-EpETE (from EPA) was positively associated with the standardized uptake value (SUV) of the TSPO-targeted radiotracer as shown in Figure 4A (P = 0.029, r = 0.4202), meaning more sequestration of this proresolving lipid mediator was linked to more inflammation. Several PL-bound oxylipins were negatively correlated with the SUV of the SV2A-targeted radiotracer, including 6 proresolving oxylipins (Figure 4B) and 4 proinflammatory oxylipins (Figure 4C). The proresolving oxylipins were DHA-derived 17-HDoHE, 19,20-DiHPDA, and 19(20)-EpDPE; EPA-derived 15-HEPE; AA-derived 15-HETE; and ALA-derived 15(S)-HETrE (r =–0.5031 to –0.3907; P = 0.008–0.044). The proinflammatory oxylipins were AA-derived 11-HETE, 9-HETE, 5-HETE, and 5,6-DiHETrE (r = –0.4359 to –0.3917, P = 0.023 to 0.043). This means that the presence of more of these oxylipin in the esterified PL form was associated with fewer synapses.
 Figure 4
Figure 4Pearson’s correlation between brain in vivo imaging markers of AD pathology and brain esterified oxylipins in female rats. The figures show linear plots and significant Pearson’s correlation coefficient r values between in vivo imaging markers of AD pathogenesis and log oxylipin concentrations within PL or NL in female rats (n = 27). (A) 18F-PBR111 average whole brain SUV and log-transformed PL-bound oxylipins. (B) 18F-UCB-H average whole brain SUV and log-transformed proresolving PL-bound oxylipins. (C) 18F-UCB-H average whole brain SUV and log-transformed proinflammatory PL-bound oxylipins. (D) 18F-FDG average whole brain SUV and log-transformed NL-bound oxylipins. *P < 0.05 and **P < 0.01. DiHETrE, dihydroxyeicosatrienoic acid; DiHDPA, dihydroxydocosapentaenoic acid; EpDPE, epoxydocosapentaenoic acid; EpETE, epoxyeicosatetraenoic acid; EpETrE, epoxyeicosatrienoic acid; HDoHE, hydroxydocosahexaenoic acid; HEPE, hydroxyeicosapentaenoic acid; HETE, hydroxyeicosatetraenoic acid; HETrE, hydroxyeicosatrienoic acid; HODE, hydroxyoctadecadienoic acid; HOTrE, hydroxyoctadecatrienoic acid.
 Table 1
Table 1Effects of genotype and TRAP exposure on brain volume and AD neuropathology markers of AD in 15-month-old female rats
There were no significant correlations between NL-bound oxylipins and SUV of the TSPO- and SV2A-targeted radiotracers. However, ALA-derived 13-HOTrE (P = 0.027, r = –0.4253) and LA-derived 9-hydroxyoctadecadienoic acid (9-HODE) (P = 0.035, r = –0.4075) in NL were negatively correlated with 18F-FDG SUV (Figure 4D), meaning greater esterified levels of these compounds were related to lower brain glucose metabolism.
Association between age at death and esterified oxylipins in human postmortem brains. We next explored whether the observed link between brain esterified oxylipins and markers of AD pathogenesis in rats translated to human postmortem prefrontal cortex of individuals with or without AD.
There were no significant associations between oxylipins and AD in NL. However, a significant age/AD interaction was observed in 11 oxylipins esterified to PL, after adjusting for postmortem interval (PMI), sex, and age at death (Table 2 and Supplemental Table 7). As shown in Figure 5, proresolving epoxides of AA [14(15)-EpETrE] (Figure 5B) and DHA [19(20)-EpDPE] (Figure 5D) decreased with age at death in individuals with AD and increased with age at death in non-AD controls. With increasing age at death, 11 PL-bound oxylipins of LOX enzymatic synthesis increased in AD but decreased in the control (non-AD) group. These included LA-derived 9-HODE and 13-HODE (Figure 5A); DGLA-derived 15(S)-HETrE (Figure 5C); AA-derived 5-HETE, 8-HETE, 11-HETE, 12-HETE, and 15-HETE (Figure 5B); and DHA-derived 17-HDoHE (Figure 5D), of which 17-HDoHE, 5-HETE, and 15-HETE are precursors to proresolving lipid mediators not detected in this study, while the other compounds are proinflammatory.
 and age in prefrontal cortex of individuals with AD and without AD.") Figure 5
Figure 5Association between PL-bound oxylipins (log-transformed values) and age in prefrontal cortex of individuals with AD and without AD. Linear plots and Pearson’s correlation coefficient (r value) between oxylipins and age in the Control (n = 20) and AD (n = 21) groups. These plots are used to visualize associations that reached statistical significance by multiple linear regression analysis applied on log-transformed concentrations of each oxylipin as the dependent variable, and group, sex, mean-centered postmortem interval (PMI), and mean centered age were tested for main effects (independent variables). Interaction effects were tested between group and age as well as between group and sex. (A–D) LA-derived oxylipins (A), DGLA-derived oxylipins (B), AA-derived oxylipins (C), and DHA-derived oxylipins (D). r values in the figure represent Pearson’s correlation coefficient of the linear plot correlating log-transformed oxylipins to age for each of the AD and non-AD groups. EpDPE, epoxydocosapentaenoic acid; EpETE, epoxyeicosatetraenoic acid; EpETrE, epoxyeicosatrienoic acid; HDoHE, hydroxydocosahexaenoic acid; HETE, hydroxyeicosatetraenoic acid; HETrE, hydroxyeicosatrienoic acid; HODE, hydroxyoctadecadienoic acid.
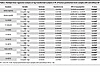 Table 2
Table 2Multiple linear regression analysis on log-transformed oxylipins in PL of human postmortem brain samples with and without AD

 female rats exposed to FA or TRAP for 14 months.")
 female rats exposed to FA or TRAP for 14 months.")


 and age in prefrontal cortex of individuals with AD and without AD.")



