Cell biology
Abstract
FGF13, a noncanonical fibroblast growth factor (FGF) and member of the fibroblast growth factor homologous factor (FHF) subset, lacks a signal sequence and was previously reported to remain intracellular, where it regulates voltage-gated sodium channels (VGSCs) at least in part through direct interaction with the cytoplasmic C-terminus of VGSCs. Recent reports suggest that FGF13 is secreted and regulates neuronal VGSCs through interactions with extracellular domains of integral plasma membrane proteins, yet supportive data are limited. Using rigorous positive and negative controls, we showed that transfected FGF13 is not secreted from cultured cells in a heterologous expression system nor is endogenous FGF13 secreted from cultured neurons. Further, employing multiple unbiased screens including proximity protein proteomics, our results suggested FGF13 remains within membranes and is unavailable to interact directly with extracellular protein domains.
Authors
Mattia Malvezzi, Haiying Zhang, Patrick Towers, David C. Lyden, Steven O. Marx, Geoffrey S. Pitt
Abstract
Ehlers-Danlos Syndrome, Classic-Like, 2 (clEDS2) is a rare genetic disorder caused by biallelic mutations in the AEBP1 gene, which encodes Aortic carboxypeptidase-like protein (ACLP). Patients with clEDS2 exhibit hallmark features such as loose connective tissues, osteoporosis, and scoliosis. Despite its clinical significance, the molecular mechanisms underlying AEBP1 mutations in skeletal development remain poorly understood, and effective therapeutic strategies are currently unavailable. Here, using OsxCre conditional knockout mice, we show that Aebp1 deletion in osteoprogenitors reduces body size and bone mass, recapitulating key skeletal features reported in clEDS2. In primary osteoblasts, both genetic deletion and siRNA-mediated knockdown of Aebp1 impair osteoblast differentiation. Mechanistically, Aebp1 loss attenuates Wnt/β-catenin signaling in bone. Restoration of Wnt/β-catenin signaling by injecting BIO, a small molecule inhibitor of GSK3, substantially rescued bone mass reduction in Aebp1 knockout mice. These findings support a model in which Aebp1 sustains baseline Wnt/β-catenin tone in osteoblast-lineage cells and suggest that Wnt-targeted approaches may help mitigate clEDS2-related skeletal defects.
Authors
Shuhao Feng, Zihang Feng, Zhonghao Deng, Yiran Wei, Ru Lian, Yangchen Jin, Shiqi Zhao, Yu Jin, Zhongmin Zhang, Liang Zhao
Abstract
Host factors influencing susceptibility to rhinovirus-induced asthma exacerbations remain poorly characterized. Using organotypic bronchial epithelial cultures from well-characterized children with asthma and healthy children, this study investigated viral load kinetics and resultant host responses by bulk and single-cell transcriptomics and targeted protein analyses. Bronchial epithelium from exacerbation-prone children exhibited greater rhinovirus replication and a cascade of exaggerated downstream interferon (IFN), inflammatory, epithelial stress, and remodeling responses. These transcriptional patterns were confirmed and further refined using single-cell transcriptomics, revealing cell type-specific contributions—particularly from non- ciliated cell populations including secretory immune response, tuft, and basal cells. We observed that these post-infection differences were associated with lower pre-infection IFN- stimulated gene (ISG) expression and protein levels of the ISG CXCL10. Prophylactic IFN-β treatment reduced viral replication and normalized downstream responses, supporting low baseline (pre-infection) IFN tone as a modifiable causal determinant of host susceptibility to adverse rhinovirus-induced responses in exacerbation-prone children with asthma.
Authors
Naresh Doni Jayavelu, Basilin Benson, Patricia C. dela Cruz, Weston T. Powell, Lucille M. Rich, Elizabeth R. Vanderwall, Camile R. Gates, Andrew J. Nagel, Maria P. White, Nyssa B. Samanas, Kourtnie Whitfield, Teal S. Hallstrand, Steven F. Ziegler, Matthew C. Altman, Jason S. Debley

Abstract
Renal polycystins (PKD1, PKD2) are ion channel–forming subunits that traffic to principal cell primary cilia. Variants in these proteins cause approximately 95% of autosomal dominant polycystic kidney disease (ADPKD), a common, lethal genetic disorder that lacks effective drug treatments. We assessed the mechanistic impact and pathogenic propensity of 2 disease-associated PKD2 truncating variants, R803X and R654X. Worldwide, hundreds of individuals with ADPKD harbor these germline mutations, including the R803X founder variant first identified within the patient population of Taiwan. Our biochemical, electrophysiological, and super-resolution imaging analyses demonstrated that the pore-truncating R654X variant abolished channel assembly and ciliary trafficking, whereas the R803X variant retained partial cilia trafficking and channel function. To assess disease impact, we generated transgenic mice with analogous truncation mutations. Homozygous mutants were embryonic lethal, whereas heterozygous mice expressing both variant and conditional Pkd2 repression alleles developed pronounced renal cysts. Cyst progression was slower in mice carrying the equivalent Taiwan mutation, reflecting the milder clinical course observed in patients. These findings revealed that the degree of impaired PKD2 channel trafficking to primary cilia correlated with cystic disease severity, providing insight into variant-specific ADPKD pathogenesis and newly developed animal models expressing clinically relevant variants for therapeutic testing.
Authors
Louise F. Kimura, Orhi Esarte Palomero, Megan Larmore, Paul G. DeCaen, Thuy N. Vien
Abstract
Chronic liver injury results in activation of quiescent Hepatic Stellate Cells (qHSCs) into Collagen Type I-producing activated HSCs that make liver fibrotic. We identified ETS1/2 (E26 transformation-specific transcription factors 1/2) as lineage-specific transcription factors regulating HSC phenotypes. Here we investigated the role of ETS1/2 in HSCs in liver fibrosis using toxic liver injury models and 3D human liver spheroids. Liver fibrosis was induced in wild-type and HSC-specific Ets1 (Ets1ΔHSC) and Ets2 (Ets2ΔHSC) knockout mice by administration of carbon tetrachloride for 6 weeks, following cessation of liver injury for 2 weeks. Liver fibrosis was more severe in Ets1ΔHSC, and to lesser extent in Ets2ΔHSC, compared to wild-type mice. Regression of liver fibrosis was suppressed only in Ets1ΔHSC, indicating Ets1 as the predominant isoform maintaining quiescent-like phenotype in HSCs. Similar results were obtained in a MASH model using 3D human liver spheroids. Knockdown of ETS1 in human HSCs caused upregulation of fibrogenic genes in MASH human liver spheroids and prevented fibrosis regression. ETS1 regulated the qHSC phenotype via CRTC2/PGC1α/PPARγ pathway. Knockdown of CRTC2 (cAMP response element-binding protein (CREB)-regulated transcription co-activator 2) abrogated PPARγ responses and facilitated HSC activation. These findings suggest that ETS1 may represent a therapeutic target for anti-fibrotic therapy.
Authors
Wonseok Lee, Xiao Liu, Sara Brin Rosenthal, Charlene Miciano, Sadatsugu Sakane, Kanani Hokutan, Debanjan Dhar, Hyun Young Kim, David A. Brenner, Tatiana Kisseleva
Abstract
Clear cell renal cell carcinomas (ccRCC) are largely driven by HIF2α and are avid consumers of glutamine. However, inhibitors of glutaminase1 (GLS1), the first step in glutaminolysis, have not shown benefit in phase III trials, and HIF2α inhibition, recently FDA-approved for treatment of ccRCC, shows significant but incomplete benefits. This highlights the need to better understand the interplay between glutamine metabolism and HIF2α in ccRCC. Here, we report that glutamine deprivation rapidly redistributes GLS1 into isolated clusters within mitochondria in diverse cell types, but not in ccRCC. GLS1 clustering occurs rapidly within 1 to 3 hours, is reversible, is specifically triggered by reduced intracellular glutamate, and is dependent on mitochondrial fission. Clustered GLS1 markedly enhances glutaminase activity and promotes cell death under glutamine-deprived conditions. HIF2α prevents GLS1 clustering, independently of its transcriptional activity, thereby maintaining low GLS activity and protecting ccRCC cells from glutamine deprivation-induced cell death. Forced clustering of GLS1, using constitutively clustering mutants, restores high GLS activity, promotes apoptosis, and suppresses ccRCC tumor growth in vivo. These findings reveal multiple insights into cellular glutamine handling, including a previously unrecognized process by which HIF2α promotes ccRCC: by suppressing GLS1 clustering and maintaining low GLS activity. This mechanism provides a potential explanation for the lack of clinical efficacy of GLS inhibitors in ccRCC and suggests a therapeutic avenue to combine HIF2α inhibition with strategies that restore GLS1 clustering.
Authors
Wencao Zhao, Sara M. Demczyszyn, Nathan J. Coffey, Yanqing Jiang, Boyoung Kim, Schuyler Bowers, Caitlyn E. Bowman, Michael C. Noji, Cholsoon Jang, M. Celeste Simon, Zoltan Arany, Boa Kim
Abstract
The lung’s mechanosensitive immune response to alveolar overdistension impedes ventilation therapy for hypoxemic respiratory failure. Though mechanistically unclear, the prevailing hypothesis is that the immune response results when alveolar overdistension stretches alveolar macrophages (AMs). Since this hypothesis is untested in live lungs, we optically imaged live mouse alveoli to detect alveolus-adherent, sessile AMs that communicate with the alveolar epithelium through connexin43 (Cx43)-containing gap junctions. Alveolar hyperinflation did not stretch the AMs, but it increased AM Ca2+. AM-specific Cx43 deletion blocked the Ca2+ response, as well lung injury due to mechanical ventilation at high tidal volume (HTV). HTV was also inhibited by AM-targeted delivery of liposomes containing the inhibitor of endosomal Ca2+ release, Xestospongin C. We conclude, Cx43- and Ca2+-dependent AM-epithelial interactions determine the lung’s mechanosensitive immunity, providing a basis for therapy for ventilator-induced lung injury.
Authors
Liberty Mthunzi, Mohammad Islam, Galina A Gusarova, Brian Karolewski, Sunita Bhattacharya, Jahar Bhattacharya
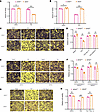
Abstract
Apoptosis and necroptosis are 2 distinct destinies of cells stimulated with TNF-α; however, it remains unclear how apoptosis and necroptosis are differentially regulated. This study validates the key regulatory role of speckle-type POZ protein (SPOP) in balancing apoptosis and necroptosis. SPOP promotes the polyubiquitination and degradation of receptor-interacting serine/threonine-protein kinase 3 (RIPK3), thereby inhibiting necrosome formation and decreasing cellular sensitivity to necroptosis. Conversely, SPOP interacted with RIPK1 independently of its E3 ubiquitin ligase activity, protecting it from ubiquitination and degradation, thereby enhancing RIPK1 expression and cellular sensitivity to apoptosis. Inhibiting RIPK1 kinase activity with 7-Cl-O-Nec-1 impeded both SPOP-mediated apoptosis and SPOP deficiency–mediated necroptosis. Besides, inhibition or loss of RIPK3 rescued SPOP deficiency–mediated necroptosis. Pancancer analyses indicated that the SPOP/RIPK1/RIPK3 axis is dysfunctional in a variety of tumors. In 3 representative tumor types with high expression of SPOP and RIPK1, kidney renal clear cell carcinoma, liver hepatocellular carcinoma, and breast invasive carcinoma, this regulatory mechanism remains applicable. Based on these findings, a combination therapy using the second mitochondria-derived activator of caspases (Smac) mimetic SM164 and sunitinib was developed, demonstrating a more pronounced efficacy than sunitinib monotherapy, and this sensitizing effect was dependent on the expression level of RIPK1. These results suggest that the combination of Smac mimetics with tyrosine kinase inhibitors holds potential clinical value for tumors with dysregulated SPOP/RIPK1/RIPK3 signaling.
Authors
Yuzhong Ye, Changjie Yue, Zaosong Zheng, Hailong Ruan, Yuanpeng Zhang, Qi Miao, Xiaoping Zhang, Wen Xiao, Lei Liu
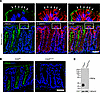
Abstract
The gastrointestinal epithelium depends on the apical junctional complex (AJC), composed of tight and adherens junctions, to regulate barrier function. Here, we identify the apical polarity protein Crumbs homolog 3 (CRB3) as an important regulator of AJC assembly and barrier function in intestinal epithelium. Using primary murine colonic epithelial cells (colonoids) from inducible, conditional Crb3-knockout (Crb3ERΔIEC) and control (Crb3fl/fl) mice, we show that CRB3 deficiency compromised barrier function that was associated with a hypercontractile perijunctional actomyosin network and impaired AJC assembly. Loss of CRB3 exacerbated proinflammatory cytokine–induced AJC remodeling, leading to increased intestinal permeability. Crb3ERΔIEC cells exhibited increased RhoA activity and junctional tension, which could be reversed by ROCK-II or myosin II inhibition, restoring junctional architecture. Mechanistically, CRB3A interacts with the actin cytoskeletal linker protein, Merlin (NF2) via its FERM-binding domain, and NF2 knockdown phenocopied CRB3 loss, suggesting their cooperative role in AJC assembly. These findings establish CRB3 and NF2 signaling as key regulators of perijunctional actomyosin contractility and AJC organization during both de novo junctional assembly and inflammation-induced remodeling. This work defines a CRB3- and NF2-dependent pathway by which epithelial cells regulate mechanical tension to coordinate barrier assembly during homeostasis and junctional remodeling under inflammatory stress.
Authors
Shuling Fan, Saranyaraajan Varadarajan, Vicky Garcia-Hernandez, Sven Flemming, Arturo Raya-Sandino, Ben Margolis, Charles A. Parkos, Asma Nusrat
Abstract
Laminin-α2-related Congenital Muscular Dystrophy (LAMA2-CMD) is a severe neuromuscular disorder caused by mutations in the LAMA2 gene, leading to loss of heterotrimers laminin-211/221, key components of the skeletal muscle extracellular matrix. Their absence disrupts adhesion between the cytoskeleton and extracellular matrix, resulting in progressive muscle wasting. Laminin-211/221 interacts with adhesion complexes such as the dystrophin/Utrophin glycoprotein complex and the α7β1-integrin. However, the regulatory mechanisms of these laminin-binding complexes and the broader role of laminin’s influence on the formation of the macromolecular network in skeletal muscle remain unclear. We previously demonstrated that mouse laminin-111 delivered in the dyW⁻/⁻ mouse model of LAMA2-CMD prevented disease progression, improved strength, and extended survival. We hypothesize that laminin-111, the embryonic laminin isoform, restores key adhesion-signaling networks. Using spatial-proteomics on patient and mouse muscle, we identified loss of essential signaling components: heat shock proteins 27 and 70, c-Jun N-terminal kinase, and glucose transporter 1 in laminin-α2 deficient muscle. Treatment with recombinant human laminin-111 (rhLAM-111) restored protein localization, reduced ROS, and promoted glycolytic, pro-survival signaling. These findings highlight laminin’s role in maintaining muscle homeostasis and metabolism and support the therapeutic potential of rhLAM-111 for treating LAMA2-CMD by restoring adhesion and intracellular signaling in dystrophic muscle.
Authors
Hailey J. Hermann, Ryan D. Wuebbles, Marisela Dagda, Axel Munoz, Lauren L. Parker, Paula C. C Guzman, Lola T. Byrne, Steven A. Moore, Dean J. Burkin
No posts were found with this tag.


