Hyperosmotic stimuli activate polycystin proteins to aid in urine concentration
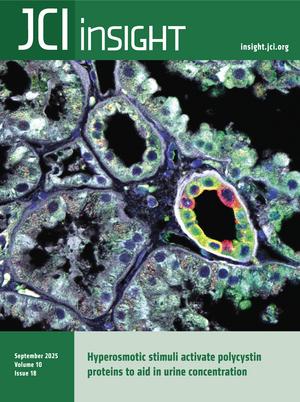
Márquez-Nogueras et al. report that the polycystin proteins, implicated in a genetic kidney disease, are essential for the kidney to concentrate urine. The cover art shows staining of aquaporin 2 (red), microtubule-associated protein 4 (MAP4, green), nuclei (blue), and actin (white) in a kidney slice from a patient diagnosed with autosomal dominant polycystic kidney disease (ADPKD), with cytosolic localization of aquaporin 2 in the collecting duct. Image credit: Karla Márquez-Nogueras.
-
Research Letters
×
Abstract
Authors
Joshua A. Keefe, Jose Alberto Navarro-Garcia, Shuai Zhao, Mihail G. Chelu, Xander H.T. Wehrens
×Abstract
Authors
Veethika Pandey, Heike R. Döppler, Ligia I. Bastea, Alicia K. Fleming Martinez, Barath Shreeder, Brandy H. Edenfield, Keith L. Knutson, DeLisa Fairweather, Peter Storz


-
Research Articles
×
Abstract
Mucous membrane pemphigoid (MMP) is a mucocutaneous autoimmune blistering disease affecting diverse mucous membranes and the skin with inflammatory blisters and erosions. The pathogenesis of MMP is only poorly understood, but inflammation in MMP is triggered by specific binding of autoantibodies directed to different proteins of the dermal-epidermal/-epithelial junction, subsequently leading to the influx of inflammatory cells, particularly neutrophils, into the dermis. Using the anti-laminin 332 antibody transfer model of MMP, we addressed the molecular mechanisms of neutrophil infiltration and its significance for the eruption of mucocutaneous lesions. Mice deficient in 5-lipoxygenase (Alox5–/–) or in the leukotriene B4 (LTB4) receptor BLT1 (Ltb4r1–/–) were resistant to skin inflammation and exhibited substantially fewer mucosal lesions, with deficiency in either gene compromising the recruitment of neutrophils to the lesion. Furthermore, neutrophil-specific genetic deficiency in Ltb4r1 similarly protected from MMP. Hence, BLT1 was required on neutrophils, and neutrophil recruitment was indispensable for the eruption of lesions in MMP. In line with these findings, the BLT1 inhibitor CP-105,606 ameliorated MMP dose-dependently. Collectively, our results highlight neutrophils and LTB4/BLT1 as key drivers of inflammation in MMP and as promising therapeutic targets.
Authors
Tabea Bremer, Sripriya Murthy, Sabrina Patzelt, Paul Schilf, Mareike Neumann, Sina Gonther, Jasper Pruessmann, Wiebke Pruessmann, Enno Schmidt, Thomas Rülicke, Christian D. Sadik
×Abstract
Glycogen storage disease type Ia (GSD Ia) is caused by a deficiency of glucose-6-phosphatase (G6Pase) in the liver leading to lethal hypoglycemia. Gene therapy with adeno-associated virus (AAV) vectors encoding G6Pase fails to stably treat GSD Ia early in life. We evaluated genome editing in 12-day-old infant mice with GSD Ia using 2 AAV vectors, one containing Cas9 from Streptococcus pyogenes and a second Donor vector that expresses a guide RNA and a G6PC transgene. Gene therapy with the Donor vector only was compared with genome editing using both Donor and CRISPR vectors. Treatment with genome editing (total vector dose 0.2 × 1013 to 2 × 1013 vector genomes/kg) and bezafibrate (to stimulate autophagy) was efficacious, as assessed by hypoglycemia prevention and the frequency of transgene integration, which correlated with improved survival. This therapy achieved 5.9% chromosomal transgene integration through homology-directed repair, which surpassed a threshold to prevent long-term hepatic complications. No integration was detected in the absence of the CRISPR vector. Importantly for safety, CRISPR vector genomes were depleted, and no intact, integrated CRISPR genomes were detected by long-read sequencing. Thus, genome editing warrants further development as a potentially stable treatment for human infants with GSD Ia.
Authors
Benjamin Arnson, Ekaterina Ilich, Troy von Beck, Songtao Li, Elizabeth D. Brooks, Dorothy Gheorghiu, Gordon He, Matthew Weinrub, Sze Ying Chan, Hye-Ri Kang, David Courtney, Jeffrey I. Everitt, Bryan R. Cullen, Dwight D. Koeberl
×Abstract
Regulatory T cells (Tregs) are essential for peripheral tolerance and depend on TCR and IL-2 receptor (IL-2R) signaling for their homeostasis and function. In mice, IL-2–dependent B-lymphocyte-induced maturation protein 1 (BLIMP-1) contributes to Treg homeostasis. BLIMP-1 is a major transcriptional hub in human Tregs, but its mechanisms of action remain undefined. Here, using CRISPR/Cas9 ablation, we show that BLIMP-1 limits human Treg proliferation but supports IL-10, cytotoxic T lymphocyte-associated protein 4, several immune checkpoints including carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), and Treg functional activity. BLIMP-1 restrains Treg expansion to IL-2 by downregulating CD25 and IL-2R signaling, and by enhancing CEACAM1 expression, which in turn inhibits responsiveness to CD3/CD28 signaling and activation of mTOR. Prolonged IL-2R signaling optimizes BLIMP-1 expression, supporting chromosomal opening of CEACAM1 to increased CEACAM1 expression through STAT5- and BLIMP-1–driven enhancers. Correspondingly, CEACAM1 is highly induced on Tregs from patients with autoimmune disease undergoing low-dose IL-2 therapy, and these Tregs showed reduced proliferation. A humanized mouse model of xenogeneic graft-versus-host disease demonstrates that BLIMP-1 normally promotes, while CEACAM1 restrains, Treg suppressive activity. Collectively, our findings reveal that BLIMP-1 and CEACAM1 function in an IL-2–dependent feedback loop to restrain Treg proliferation and affect suppressive function. CEACAM1 also acts as a highly selective biomarker of IL-2R signaling in human T cells.
Authors
Ying Ding, Aixin Yu, Milos Vujanac, Sabrina N. Copsel, Alejandro Moro, Luis Nivelo, Molly Dalzell, Nicolas Tchitchek, Michelle Rosenzwajg, Alejandro V. Villarino, Robert B. Levy, David Klatzmann, Thomas R. Malek
×Abstract
Pathological cardiac remodeling is associated with the reactivation of fetal genes, yet the extent of the heart’s fetal gene program and its impact on proteome compositions remain incompletely understood. Here, using a proteome-wide protein ratio quantification strategy with mass spectrometry, we identified pervasive isoform usage shifts in fetal and postnatal mouse hearts, involving 145 pairs of highly homologous paralogs and alternative splicing–derived isoform proteins. Proteome-wide ratio comparisons readily rediscovered hallmark fetal gene signatures in muscle contraction and glucose metabolism pathways, while revealing what we believe to be previously undescribed isoform usage in mitochondrial and gene-expression-regulating proteins, including PPA1/PPA2, ANT1/ANT2, and PCBP1/PCBP2 switches. Paralogs with differential fetal usage tend to be evolutionarily recent, consistent with functional diversification. Alternative splicing adds another rich source of fetal isoform usage differences, involving PKM M1/M2, GLS1 KGA/GAC, PDLIM5 long/short, and other spliceoforms. When comparing absolute protein proportions, we observed a partial reversion toward fetal gene usage in pathological hearts. In summary, we present a ratiometric catalog of paralogs and spliceoform pairs in the cardiac fetal gene program. More generally, the results demonstrate the potential of applying the proteome-wide ratio test concept to discover new regulatory modalities beyond differential gene expression.
Authors
Yu Han, Shaonil Binti, Sara A. Wennersten, Boomathi Pandi, Dominic C.M. Ng, Edward Lau, Maggie P.Y. Lam
×Abstract
Reprogramming autoreactive CD4+ effector T (Teff) cells into immunosuppressive Tregs represents a promising strategy for treating established autoimmune diseases. However, the stability and function of such reprogrammed Tregs under inflammatory conditions remain unclear. Here, we show that demethylation of core Treg identity genes in Teff cells yields lineage-stable effector T cell reprogrammed Tregs (ER-Tregs). A single adoptive transfer of ER-Tregs not only prevents autoimmune neuroinflammation in mice when given before disease onset but also arrests its progression when administered after onset. Compared with Foxp3-overexpressing Teff cells, induced Tregs from naive precursors, and endogenous Tregs, ER-Tregs provide superior protection against autoimmune neuroinflammation. This enhanced efficacy stems from their inherited autoantigen specificity and selectively preserved effector cell transcriptional programs, which together bolster their fitness in inflammatory environments and enhance their suppressive capacity. Our results establish epigenetic reprogramming of autoreactive Teff cells as an effective approach to generate potent, stable Tregs for the treatment of refractory autoimmune conditions.
Authors
Tyler R. Colson, James J. Cameron, Hayley I. Muendlein, Mei-An Nolan, Jamie L. Leiriao, James H. Kim, Alexander N. Poltorak, Xudong Li
×Abstract
Infections with nontuberculous mycobacterium (NTM) are on the rise. Here, we investigated an uncommon NTM infection, by M. haemophilum (Mh, n = 4), from a shared geographic location in the United States. All patients had underlying immunosuppressive conditions or treatments. We identified that all these individuals had a nonsynonymous mutation in GATA2 gene, which was absent in healthy controls (HCs, n = 4) from the same geographic area (Missouri, USA). Whole blood from these individuals had attenuated cytokine responses to Mh stimulation for IL-1β, IL-6, IL-8, MIP-1α and MIP-1β, but not to phytohemagglutinin (PHA) or another NTM, M. abscessus. Impaired whole blood transcriptional responses in individuals with GATA2 mutation included heightened Ras-homolog (Rho) guanosine triphosphate hydrolases (GTPase) and lowered TGF-β responses, among others. Our results highlight that, comparatively, M. abscessus and Mh elicit differential immune responses in humans. We identify a 23-gene signature that distinguished host response to Mh and M. abscessus and show that in vitro GATA2 siRNA knockdown indeed attenuated cytokine responses to Mh. Thus, we provide evidence that links GATA2 mutation and immune dysfunction in individuals with compromised immunity to Mh infection in humans and outline host factors associated with the immune response of this clinically relevant NTM.
Authors
Ananya Gupta, Shail B. Mehta, Abhimanyu, Bruce A. Rosa, John Martin, Mushtaq Ahmed, Shyamala Thirunavukkarasu, Farheen Fatma, Gaya K. Amarasinghe, Makedonka Mitreva, Thomas C. Bailey, David B. Clifford, Shabaana A. Khader
×Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in PKD1 or PKD2, which encode polycystin-1 (PC1) and polycystin-2 (PC2), respectively. These proteins are thought to form a signaling complex that can flux cations, including calcium. One of the earliest symptoms in ADPKD is a decline in the concentrating ability of the kidneys, occurring prior to cyst formation. We reasoned that hyperosmolality stimulates the polycystin complex, and that the loss of this function impairs water reabsorption. We found that hyperosmolality resulted in the phosphorylation of microtubule-associated protein 4 (MAP4) in a PC1-dependent manner, which then elicited ER-localized PC2 calcium signals. ER-localized PC2 hyperosmotic calcium signals were required for trafficking of the water channel aquaporin (AQP2). Precystic PC1-KO and PC2-KO murine kidneys had cytosol-localized AQP2 and diluted urine compared with their respective controls. Kidney tissue sections from ADPKD patients showed decreased AQP2 apical membrane localization in cystic and noncystic tubules. Our study demonstrates that osmolality is a physiological stimulus of the polycystin complex, and loss of polycystin osmosensing results in impaired water reabsorption via AQP2. This likely contributes to the declined concentrating ability of the kidneys and high circulating vasopressin levels in patients with ADPKD.
Authors
Karla M. Márquez-Nogueras, Ryne M. Knutila, Virdjinija Vuchkovska, Charlie Yang, Patricia Outeda, Darren P. Wallace, Ivana Y. Kuo
×Abstract
Secreted high mobility group box protein 1 (HMGB1) regulates the adaptive immune response and acts as a biosensor for cells undergoing necrosis, stress, and inflammatory stimulation. However, its role in B cells remains enigmatic. Here, we demonstrate that HMGB1 is critical for peripheral B cell homeostasis and humoral immunity. Conditional deletion of Hmgb1 in B cells led to expanded marginal zone B cells, reduced B1a cells, and impaired antigen-specific antibody responses. Mechanistically, HMGB1 deficiency enhanced proximal and distal B cell receptor (BCR) signaling, probably via increased CD21 expression, which lowered the BCR activation threshold. This phenotype was linked to reduced lymphoid enhancer-binding factor 1 (LEF1) levels, a Wnt-responsive transcription factor, as HMGB1 directly bound the Lef1 promoter to sustain its transcription, thereby repressing Cd21. Furthermore, HMGB1 constrained actin reorganization by suppressing the MST1/DOCK8/WASP axis, which feedback-modulated BCR clustering and signalosome recruitment. Collectively, HMGB1 ensures optimal BCR signaling by transcriptionally and cytoskeletally tuning activation thresholds, highlighting its dual role as a nuclear regulator and cytoskeletal modulator in B cell immunity.
Authors
Qiuyue Chen, Ziyin Zhang, Nanshu Xiang, Li Luo, Xin Dai, Danqing Kang, Lu Yang, Yingzi Zhu, Jiang Chang, Yukai Jing, Na Li, Qianglin Chen, Panpan Jiang, Ju Liu, Yanmei Huang, Heather Miller, Xinyuan Zhou, Fang Zheng, Quan Gong, Chaohong Liu
×Abstract
Recent experimental and epidemiologic data have strongly associated air pollution in the pathogenesis of insulin resistance and type 2 diabetes mellitus. We explored the effect of inhalational exposure to concentrated ambient particulate matter smaller than 2.5 μm (PM2.5), or filtered air, using a whole-body inhalation system (6 hours/day, 5 days/week) for 24 weeks on metabolism and brown adipose tissue (BAT) function. Mechanistic evaluation of insulin resistance, glucose uptake with 18F-fluorodeoxyglucose positron emission tomography, alongside evaluation for differentially methylated regions, chromatin accessibility, and differential expression of genes was performed. PM2.5 exposure impaired metabolism through changes in key BAT transcriptional programs involved in redox stress, lipid deposition, fibrosis, and altered thermogenesis. Significant differential methylation and widespread chromatin remodeling was noted in BAT with PM2.5. Integrated analysis uncovered a role for the histone deacetylase HDAC9 and histone demethylase KDM2B. The latter demethylates Lys-4 and Lys-36 of histone H3. Specifically, studies using ChIP combined with quantitative PCR confirmed HDAC9 and KDM2B occupancy and reduced H3K36me2 on the promoter of target BAT genes in PM2.5 mice, while Hdac9/Kdm2b knockdown and overexpression increased and reduced BAT metabolism, respectively. Collectively, our results provide insights into air pollution exposure and changes in BAT and metabolism.
Authors
Rengasamy Palanivel, Jean-Eudes Dazard, Bongsoo Park, Sarah Costantino, Skanda T. Moorthy, Armando Vergara-Martel, Elaine Ann Cara, Jonnelle Edwards-Glenn, Shyam Biswal, Lung Chi Chen, Mukesh K. Jain, Francesco Paneni, Sanjay Rajagopalan
×Abstract
Anemia is a common and disabling complication of chronic kidney disease (CKD). Current therapies can be burdensome, and full correction of anemia is limited by their cardiovascular side effects. New approaches that may offer additional therapeutic options are needed. We explored the antianemic effects of erythroferrone, an erythroid hormone that induces iron mobilization by suppressing the master iron-regulatory hormone hepcidin. In a preclinical murine model of adenine-induced CKD, transgenic augmentation of erythroferrone mobilized iron, increased hemoglobin concentrations by approximately 2 g/dL, and modestly improved renal function without affecting systemic or renal inflammation, fibrosis, or markers of mineral metabolism. This study supports the concept that therapeutic augmentation of erythroferrone is a promising approach for alleviating CKD-associated anemia.
Authors
Brian Czaya, Joseph D. Olivera, Moya Zhang, Amber Lundin, Christian D. Castro, Grace Jung, Mark R. Hanudel, Elizabeta Nemeth, Tomas Ganz
×Abstract
Radiotherapy triggers chemokine release and leukocyte infiltration in preclinical models through activation of the senescence-associated secretory phenotype (SASP). However, effects of irradiation on senescence and SASP in human tissue and in the context of particle radiotherapy remain unclear. Here, we analyzed chemokine patterns after radiotherapy of human pancreatic tumors and cancer cell lines. We show that irradiated tumor cells coexpressed SASP chemokines in defined modules. These chemokine modules correlated with infiltration of distinct leukocyte subtypes expressing cognate receptors. We developed a patient-derived pancreatic tumor explant system, which verified our identified radiation-induced chemokine modules. Chemokine modules were partially conserved in cancer cells in response to photon and particle irradiation, showing a dose-dependent plateau effect, and induced subsequent migration of NK and T cell populations. Hence, our work reveals redundant interactions of cancer cells and immune cells in human tissue, suggesting that targeting multiple chemokines is required to efficiently perturb leukocyte infiltration after photon or particle radiotherapy.
Authors
Joscha A. Kraske, Michael M. Allers, Aleksei Smirnov, Bénédicte Lenoir, Azaz Ahmed, Meggy Suarez-Carmona, Mareike Hampel, Damir Krunic, Alexandra Tietz-Dalfuß, Tizian Beikert, Jonathan M. Schneeweiss, Stephan Brons, Dorothee Albrecht, Thuy Trinh, Muzi Liu, Nathalia A. Giese, Christin Glowa, Jakob Liermann, Ramon Lopez Perez, Dirk Jäger, Jürgen Debus, Niels Halama, Peter E. Huber, Thomas Walle
×Abstract
Normothermic machine perfusion (NMP) has become a valuable tool to expand the pool of transplantable organs. However, the application of NMP to kidneys presents substantial challenges, mostly due to high variability in the composition of currently used perfusion solutions. Here, we provide a multimodal cross-species cellular atlas of kidney injury associated with NMP using a literature-based consensus buffer. This resource provided a systematic framework that was used to develop a metabolite-enhanced perfusion solution, which protected renal proximal tubular cells, improving cellular viability and transplantation outcomes across species, including human kidneys.
Authors
Jan Czogalla, Fabian Hausmann, Simon Lagies, Sydney E. Gies, Sabrina Christiansen, Nico Kaiser, Fabian Haas, Yusuke Okabayashi, Dominik Kylies, Smilla Hofmann, Rossana Franzin, Niklas Sabra, Sarah Bouari, Yitian Fang, Gisela Ambagtsheer, Ilka Edenhofer, Silvia Chilla, Anne K. Mühlig, Marina Zimmermann, Milagros N. Wong, Takashi Yokoo, Oliver Kretz, Maja Lindenmeyer, Florian Grahammer, Martin J. Hoogduijn, Ron de Bruin, Malte Kuehl, Sonja Hänzelmann, Bernd Kammerer, Loreto Gesualdo, Stefan Bonn, Robert C. Minnee, Tobias B. Huber, Victor G. Puelles
×Abstract
Immune checkpoint therapy has changed cancer treatment, including non-small cell lung cancer (NSCLC). The unresponsiveness of PD-L1lo/– tumors to anti–PD-1/PD-L1 immunotherapy is attributed to alternative immune evasion mechanisms that remain elusive. We previously reported that farnesoid X receptor (FXR) was increased in PD-L1lo/– NSCLC. Herein, we found that immune checkpoint HVEM was positively correlated with FXR but inversely correlated with PD-L1 in NSCLC. HVEM was highly expressed in FXRhiPD-L1lo NSCLC. Consistently, clinically relevant FXR antagonist dose-dependently inhibited HVEM expression in NSCLC. FXR inhibited cytokine production and cytotoxicity of cocultured CD8+ T cells in vitro, and it shaped an immunosuppressive tumor microenvironment (TME) in mouse tumors in vivo through the HVEM/BTLA pathway. Clinical investigations show that the FXR/HVEM axis was associated with immunoevasive TME and inferior survival outcomes in patients with NSCLC. Mechanistically, FXR upregulated HVEM via transcriptional activation, intracellular Akt, Erk1/2 and STAT3 signals, and G1/S cycle progression in NSCLC cells. In vivo treatment experiments demonstrated that anti-BTLA immunotherapy reinvigorated antitumor immunity in TME, resulting in enhanced tumor inhibition and survival improvement in FXRhiPD-L1lo mouse Lewis lung carcinomas. In summary, our findings establish the FXR/HVEM axis as an immune evasion mechanism in PD-L1lo/– NSCLC, providing translational implications for future immunotherapy in this subgroup of patients.
Authors
Xiaolong Xu, Bin Shang, Hancheng Wu, Xiuye Jin, Junren Wang, Jing Li, Daowei Li, Bin Liang, Xingguang Wang, Lili Su, Wenjie You, Shujuan Jiang
×Abstract
Cranial neural crest cells (CNCs) play a critical role in craniofacial bone morphogenesis, engaging in intricate interactions with various molecular signals to ensure proper development, yet the molecular scaffolds coordinating these processes remain incompletely defined. Here, we identify neurofibromin 2 (Nf2) as a critical regulator to direct CNC-derived skull morphogenesis. Genetic ablation of Nf2 in murine CNCs causes severe craniofacial anomalies, featuring declined proliferation and increased apoptosis in osteoprogenitors, impaired type I collagen biosynthesis and trafficking, and aberrant osteogenic mineralization. Mechanistically, we uncover that Nf2 serves as a molecular linker that individually interacts with FGF receptor 1 (FGFR1) and Akt through spatially segregated phosphor-sites, and structural modeling and mutagenesis identified Ser10 and Thr230 as essential residues, with Thr230 mutation selectively ablating Akt binding while preserving FGFR1 association. Strikingly, Akt inhibition phenocopied Nf2 deficiency, reducing collagen production and Nf2 phosphorylation, whereas phospho-mimetic Nf2 (T230D) rescued CNC-derived osteogenic defects in Nf2-mutant animals. Our findings underscore the physiological significance of Nf2 as a phosphorylation-operated scaffold licensing the FGFR1/AKT axis to regulate collagen type I biogenesis and trafficking, ensuring normal CNC-derived osteogenesis and craniofacial bone development, thus exposing the Nf2/FGFR1/AKT signaling axis as a therapeutic target and promising advancements in treatment of craniofacial anomalies.
Authors
Yuping Huang, Junguang Liao, Panpan Shen, Yiliang He, Fuju Sun, Qi Zhang, Changlin Zheng, Xingen Zhang, Haibo Li, Guiqian Chen
×Abstract
Tumor suppressor NF1 is recurrently mutated in glioblastoma, leading to aberrant activation of Ras/rapidly accelerated fibrosarcoma (RAF)/MEK signaling. However, how tumor heterogeneity shapes the molecular landscape and efficacy of targeted therapies remains unclear. Here, we combined bulk and single-cell genomics of human somatic NF1-mutant, isocitrate dehydrogenase (IDH) wild-type glioblastomas with functional studies in cell lines and mouse intracranial tumor models to identify mechanisms of tumor heterogeneity underlying clinical outcome and MEK inhibitor response. Targeted DNA sequencing identified CDKN2A/B homozygous deletion as a poor prognostic marker in somatic NF1-mutant, but not NF1 wild-type, glioblastoma. Single-nucleus RNA sequencing of human patient NF1-mutant glioblastomas demonstrated that mesenchymal-like (MES-like) tumor cells were enriched for MEK activation signatures. Single-cell RNA-sequencing of mouse intracranial glioblastomas treated with the MEK inhibitor selumetinib identified distinct responses among tumor subpopulations. MEK inhibition selectively depleted MES-like cells, and selumetinib-resistant MES-like cells upregulated Ras signaling while resistant non-MES cells expressed markers of glial differentiation. Finally, genome-wide CRISPR interference screens validated Ras/RAF/MEK signaling as a key mediator of selumetinib response. Repression of the RAF regulator SHOC2 sensitized glioblastomas to selumetinib in vitro and in vivo, suggesting a synergistic treatment strategy. Taken together, these results highlighted the heterogeneity of NF1-mutant glioblastomas and informed future combination therapies.
Authors
Sixuan Pan, Kanish Mirchia, Emily Payne, S. John Liu, Nadeem Al-Adli, Zain Peeran, Poojan Shukla, Jacob S. Young, Rohit Gupta, Jasper Wu, Joanna Pak, Tomoko Ozawa, Brian Na, Alyssa T. Reddy, Steve E. Braunstein, Joanna J. Phillips, Susan Chang, David A. Solomon, Arie Perry, David R. Raleigh, Mitchel S. Berger, Adam R. Abate, Harish N. Vasudevan
×Abstract
Palmoplantar pustulosis (PPP) is a chronic inflammatory skin disorder marked by erythematous pustules and desquamation on the palms and soles. While IL-17 pathways are implicated in PPP, IL-17 blockers have shown modest efficacy, underscoring the need for a deeper understanding of IL-17 involvement. To dissect the cellular and spatial architecture of PPP, we performed single-cell RNA-Seq (scRNA-Seq) on lesional, nonlesional, and healthy acral skin to examine cellular composition, transcriptomic profiles, and cell-cell interactions. Unbiased clustering revealed 9 major cell types, including an inflammatory keratinocyte subset enriched in IL-17A/TNF signatures and marked by high IL-36G expression. Within the lymphocyte compartment, we identified a hybrid “regTh17” population coexpressing regulatory markers (FOXP3, CTLA4, TIGIT), IL17F, and IL26. This regTh17 subset was distinguished by elevated IL1R1 and CD39, suggesting an IL-1β–driven differentiation. Spatial analyses demonstrated significant neighborhood enrichment of regTh17 cells with IL-36G+ supraspinous keratinocytes. RegTh17 cells were the predominant source of IL-17F and IL-26 signals, whereas keratinocytes were predicted as their main receivers. We further observed regTh17 coexpressing TNFRSF4 (OX40) and TNFRSF18 (GITR) specifically at sites of IL36G+ keratinocyte interactions, implicating these pathways in amplification of the IL-17/IL-36 inflammatory loop. Together, our integrated single-cell and spatial profiling uncovers Th17 plasticity in PPP, identifies a regTh17-keratinocyte interaction, and highlights IL-17F, IL-26, OX40/OX40L, and GITR/GITRL as candidate targets for precision therapies in this challenging disease.
Authors
Tran H. Do, Rachael Bogle, Haihan Zhang, Xianying Xing, Mehrnaz Gharaee-Kermani, Madalina Raducu, Jennifer Fox, Rundong Jiang, Olesya Plazyo, Paul W. Harms, Mio Nakamura, Enze Xing, Michel Gilliet, Allison C. Billi, J. Michelle Kahlenberg, Robert L. Modlin, Ozge Uluckan, Lam C. Tsoi, Johann E. Gudjonsson
×Abstract
Radiation-induced lymphopenia (RIL) remains a challenging side effect of radiation therapy that is often associated with poor prognosis and reduced overall survival. Although CD8+ T cells are highly radiosensitive, the dynamics of quantitative and qualitative changes to the CD8+ T cell pool following exposure to high doses of ionizing radiation (IR) remain understudied. Herein, we sought to determine the long-term impact of sublethal whole body irradiation (WBI) on the antigen-inexperienced (Ag-inexperienced) CD8+ T cell pool, comprising naive (TN) and virtual memory (TVM) CD8+ T cells. We show that although both TN and TVM cells gradually regenerated after WBI-induced loss, TN recovery occurred only through de novo thymic production. Despite the numerical restoration, the subset and phenotypic composition of postrecovery Ag-inexperienced CD8+ T cells did not qualitatively recapitulate the pre-WBI state. Specifically, the frequency of TVM cells is increased, especially during the early stages of recovery. Within the TN subset, a lasting overrepresentation of Ly6C+CD122+ cells and an altered TCR clonotype diversity are also observed. Overall, our data highlight the dynamic changes to the Ag-inexperienced CD8+ T cell pool upon recovery from RIL
Authors
Mohammad Heidarian, Shravan K. Kannan, Whitney Swanson, Thomas S. Griffith, John T. Harty, Vladimir P. Badovinac
×Abstract
Hepatic ischemia-reperfusion injury (IRI) disrupts cellular signaling pathways and contributes to early allograft dysfunction (EAD) in orthotopic liver transplantation (OLT). In this study, we found that the hepatic RNA binding protein Human Antigen R (HuR) regulated the 3′ untranslated region (UTR) of Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 (Ceacam1) following ischemic stress. Hepatocyte-specific preinjury HuR-null mice exhibited elevated LDH-5 isoenzyme activity and reduced Ceacam1-S expression, reflecting tissue-specific injury. In situ hybridization demonstrated that the stability of Ceacam1 mRNA depended on HuR. Luciferase assays identified Ceacam1 3′UTR cis-elements responsive to high oxygen tension. HuR-targeting short-activating RNAs (saRNAs) preferentially induced the alternative splicing of Ceacam1-S. Antisense oligos directed to the Ceacam1 3′UTR protected WT mice against acute liver injury. In the clinical arm, increased HuR and CEACAM1 expression were associated with reduced proinflammatory phenotype and a lower incidence of EAD in patients with OLT (n = 164). Human discarded livers with elevated ELAVL1/CEACAM1 levels correlated with improved tissue homeostasis. These findings suggest that HuR regulation of Ceacam1 represents a key determinant of donor tissue quality and offers a potential target for future therapeutic strategies in OLT recipients.
Authors
Brian Cheng, Tristan D. Tibbe, Siyuan Yao, Megan Wei, Zeriel Y. Wong, Taylor Torgerson, Richard Chiu, Aanchal S. Kasargod, Kojiro Nakamura, Monica Cappelletti, Myung Sim, Douglas G. Farmer, Fady Kaldas, Jerzy W. Kupiec-Weglinski, Kenneth J. Dery
×Abstract
BACKGROUND Glucagon-like peptide-2 (GLP-2) analogs are used clinically to enhance nutrient absorption in patients with short bowel syndrome (SBS); however, the precise mechanism remains unclear. To address this, the study aimed to clarify the dynamics of intestinal epithelial cells and immune cells in patients with SBS treated with GLP-2 analogs.METHODS Five male patients diagnosed with SBS, all of whom received treatment with the GLP-2 analog teduglutide, were included in the study. We conducted longitudinal single-cell RNA sequencing (scRNA-Seq) analysis of intestinal tissue from patients with SBS over a year, integrating microbiome composition analysis.RESULTS After treatment, the α-diversity of the gut microbiome increased, indicating a more varied microbial environment. ScRNA-Seq analysis revealed a reduction of T helper 2 cells and an increase in regulatory T cells, suggesting a shift toward an immunoregulatory intestinal environment. Additionally, nutrient-absorbing enterocyte-Top2 and middle clusters expanded, enhancing the absorption capacity, whereas major histocompatibility complex class I/II–expressing enterocyte-Top1 cells declined, potentially modulating immune responses.CONCLUSION The study findings indicate that GLP-2 analogs reshape intestinal immunity and microbiota, fostering a less inflammatory environment while promoting nutrient uptake efficiency. These insights offer a deeper understanding of the role of GLP-2 analogs in gut adaptation and provide a foundation for refining clinical strategies for SBS treatment.FUNDING This work was supported by Sakaguchi Memorial Foundation, Grants-in-Aid from the Japanese Society for the Promotion of Science (JSPS) (21K18272, 23H03665, 23H02899, 23K27590, 25K22627, 23K08037), JST FOREST(21457195), and the Takeda Japan Medical Office Funded Research Grant 2022.
Authors
Yumi Kudo, Kentaro Miyamoto, Shohei Suzuki, Akihiko Chida, Anna Tojo, Mai Hasegawa, Arina Shigehara, Ikuko Koya, Yoshinari Ando, Masayasu Sato, Aya Kondo, Tomoko Kumagai, Harunori Deguchi, Yoshiki Sugiyama, Yoko Ito, Koji Shirosaki, Satoko Yamagishi, Yutaro Maeda, Hiroki Kanamori, Motohiro Kano, Mototoshi Kato, Hanako Tsujikawa, Yusuke Yoshimatsu, Kaoru Takabayashi, Koji Okabayashi, Takanori Kanai, Naoki Hosoe, Motohiko Kato, Jonathan Moody, Chung-Chau Hon, Tatsuo Kuroda, Yohei Yamada, Akihiro Fujino, Tomohisa Sujino
×Abstract
BACKGROUND. Urine proteomics may provide mechanistic insights on why patients experience a higher risk of kidney function decline after hospitalization. METHODS. In 174 patients with and without acute kidney injury (AKI) from the Assessment, Serial Evaluation, and Subsequent Sequelae in AKI (ASSESS-AKI) cohort, we used Olink to profile 2783 urinary proteins collected at 3 months after hospitalization and determined their association with estimated glomerular filtration rate (eGFR) decline during median [IQR] of 5.1 [4.0 to 6.0] years follow-up. In 4 independent cohorts, including the Kidney Precision Medicine Project (KPMP), we determined whether proteins were differentially expressed with AKI. We used weighted correlation network analysis to determine proteins’ cellular enrichment in the kidney transcriptome (single-cell and spatial transcriptomics) in patients with AKI receiving research kidney biopsy.RESULTS. We identified 387 and 10 proteins associated with faster and slower eGFR decline, respectively, most of which were differentially expressed in patients at the time of AKI. Among these proteins, 283 (71%) were expressed by kidney cells in participants with AKI from KPMP. The expression formed 3 clusters enriched in the proximal tubule, degenerative tubule and myeloid cells, and stromal cells, and correlated with histopathological features of AKI, such as tubular injury, interstitial inflammation, and fibrosis, respectively.CONCLUSION. Urinary proteins reflecting degenerative tubular injury, inflammation, and fibrosis are associated with eGFR decline in recently hospitalized patients.FUNDING. National Institute of Diabetes and Digestive Kidney Diseases grants U01DK133081, U01DK133091, U01DK133092, U01DK133093, U01DK133095, U01DK133097, U01DK114866, U01DK114908, U01DK133090, U01DK133113, U01DK133766, U01DK133768, U01DK114907, U01DK114920, U01DK114923, U01DK114933, U24DK114886, UH3DK114926, UH3DK114861, UH3DK114915, UH3DK114937, K23DK128358, R01DK128087, and R01DK140717.
Authors
Yumeng Wen, Steven Menez, Heather Thiessen Philbrook, Dennis Moledina, Steven G. Coca, Jiashu Xue, James Kaufman, Vernon Chinchillil, Paul L. Kimmel, T. Alp Ikizler, Chi-Yuan Hsu, Tanika Kelly, Ana Ricardo, Jonathan Himmelfarb, Chirag R. Parikh, ASSESS-AKI, TRIBE-AKI, and Kidney Precision Medicine Project consortia
×Abstract
There are 2 subtypes of myotonic dystrophy, DM1 and DM2, each caused by repeat expansion mutations. The leading pathogenic mechanism is RNA-mediated toxicity, whereby (C)CUG expansions sequester the muscleblind-like (MBNL) family of RNA binding proteins. However, key differences exist in muscle involvement patterns and histopathology between DM1 and DM2. The cause of these disparities both in how the muscles are affected within each disease and between the 2 diseases is unknown, and it is unclear if current DM mouse models recapitulate these differences or develop differential muscle susceptibility. Here, we examined the expression of disease-relevant genes across healthy human muscles from a transcriptomic atlas and collected a series of muscles from Mbnl-KO mice to evaluate characteristic histologic and molecular features of DM pathology. Our results indicate that MBNL loss discordantly affects muscles, likely through a splicing-independent mechanism, and results in a fiber atrophy profile more like DM1 than DM2. These findings point to a predominant role for MBNL loss in muscle pattern involvement in DM1, provide further evidence for additional DM2 pathomechanisms, and have important implications for muscle choice when performing analyses in new mouse models and evaluating therapeutic modalities and biomarkers.
Authors
Mackenzie L. Davenport, Amaya Fong, Gloria Montoya-Vazquez, Maria Fernanda Alves de Moura, Jodi L. Bubenik, Maurice S. Swanson
×Abstract
FOXP3+ Treg cells are critical for immune tolerance. Genetic deletion of the Forkhead domain–containing proteins of the FOXP-subfamily member FOXP1 from Tregs results in impaired function associated with reduced CD25 expression and IL-2 signaling, but to date the only other FOXP family member expressed in Tregs, FOXP4, has been minimally studied. To investigate the potential functional interactions among FOXP family members in Tregs, we specifically deleted Foxp1, Foxp4, or both in FOXP3+ committed Tregs in mice. Our findings show that mice with combined, but not individual, deficiency in FOXP1 and FOXP4 exhibit lymphoproliferation, inflammation, autoimmunity, and early lethality. The combined absence of FOXP1 and FOXP4 in Tregs results in an activated/effector-like phenotype with compromised suppressive function in peripheral lymphoid organs, an enhanced germinal center response, and proinflammatory cytokine production. We further show that FOXP1 and FOXP4 bind to Il2ra promoter regions to regulate CD25 expression in Tregs. Through pairwise comparison among mouse strains with Treg-specific deletion of Foxp1, Foxp4, or both, our findings indicate a nonredundant but insufficient role of FOXP4 in Treg function.
Authors
Dachuan Dong, Vishal J. Sindhava, Ananthakrishnan Ganesan, Martin S. Naradikian, Tom L. Stephen, Andrew Frisch, Kristen M. Valentine, Elizabeth Buza, Karla R. Wiehagen, Michael P. Cancro, Edward E. Morrisey, Haley Tucker, Katrina K. Hoyer, Purvesh Khatri, Jonathan S. Maltzman